

Evolutionary rescue of phosphomannomutase deficiency in yeast models of human disease
- PMID: 36214454
- PMCID: PMC9578706
- DOI: 10.7554/eLife.79346
Evolutionary rescue of phosphomannomutase deficiency in yeast models of human disease
Abstract
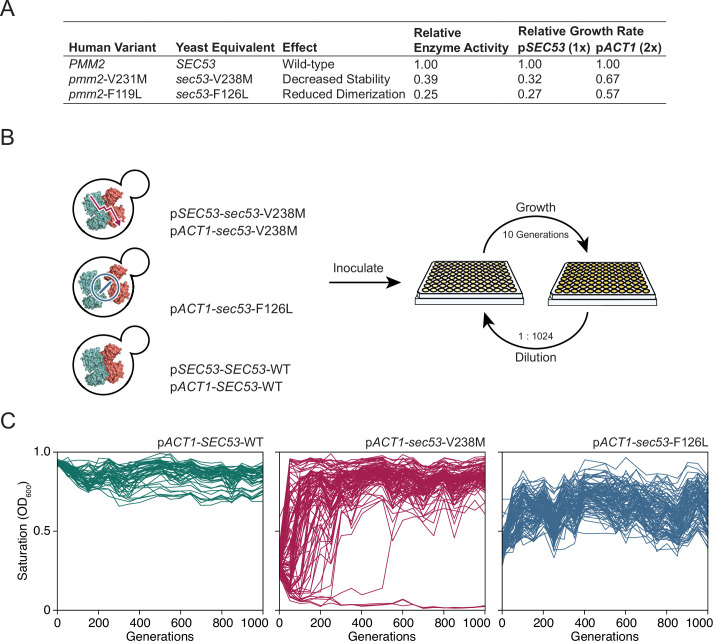
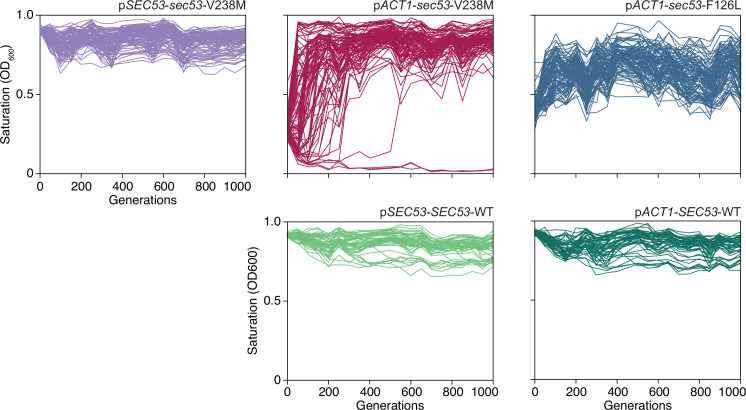
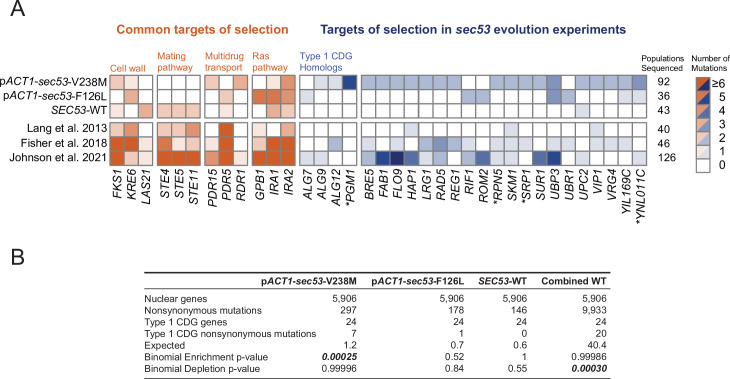
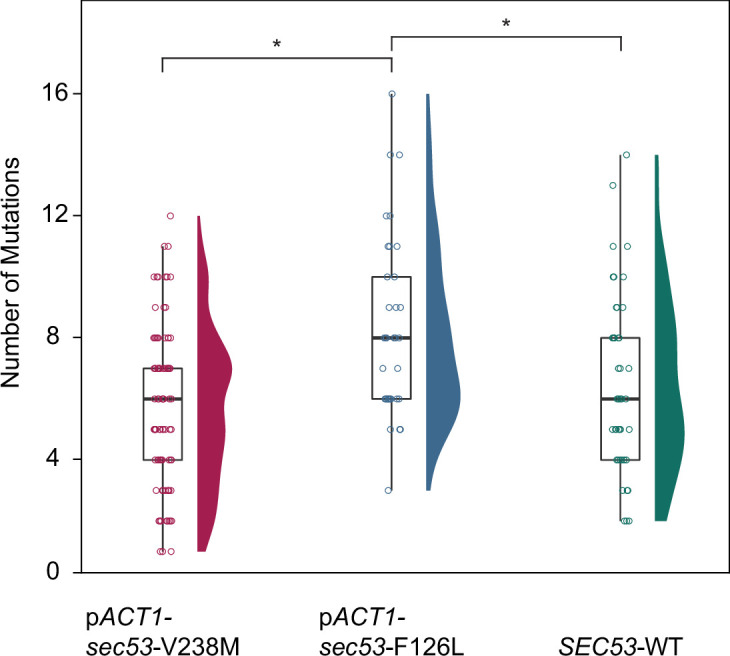
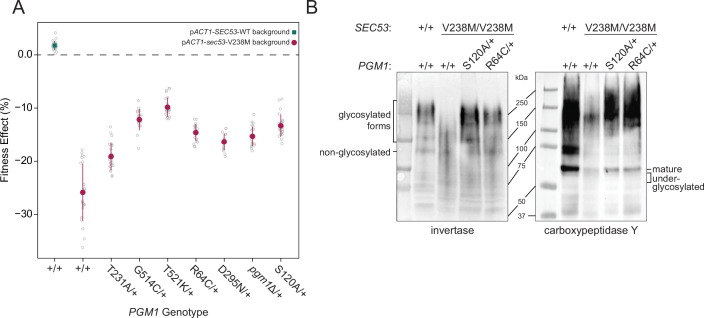
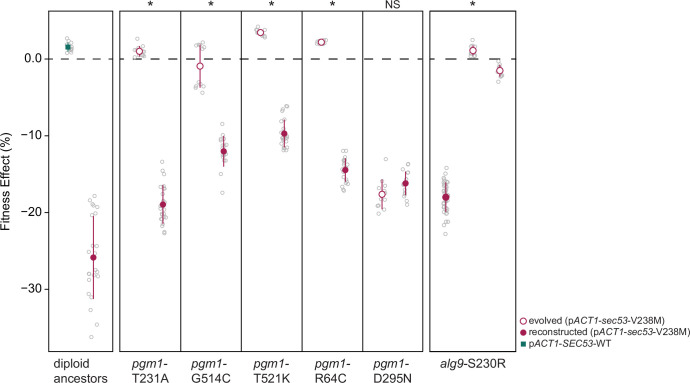
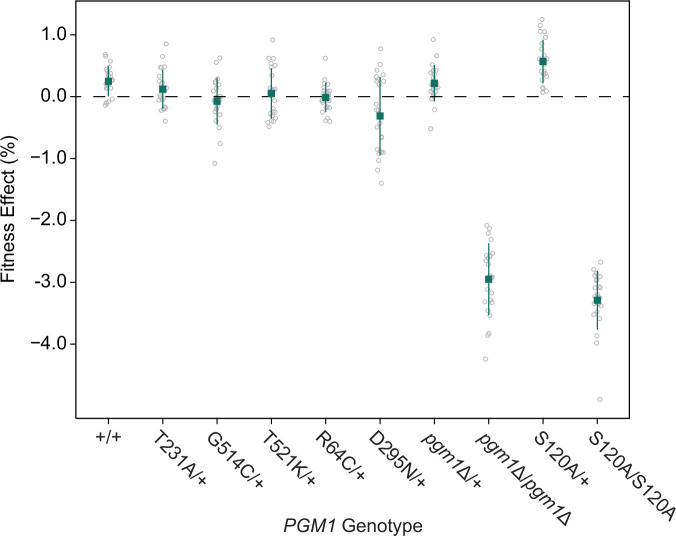
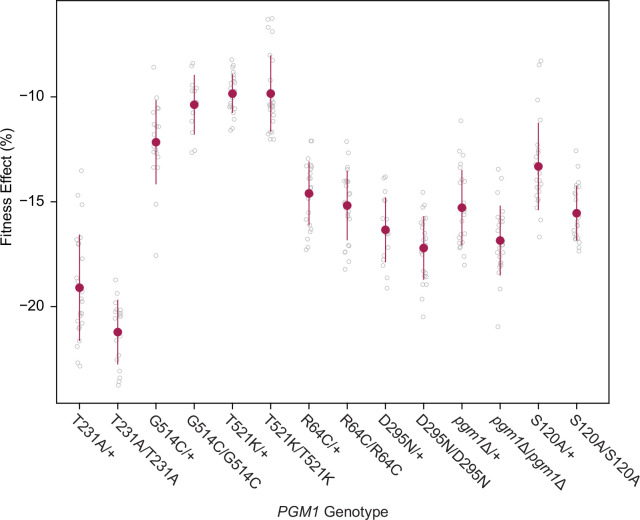
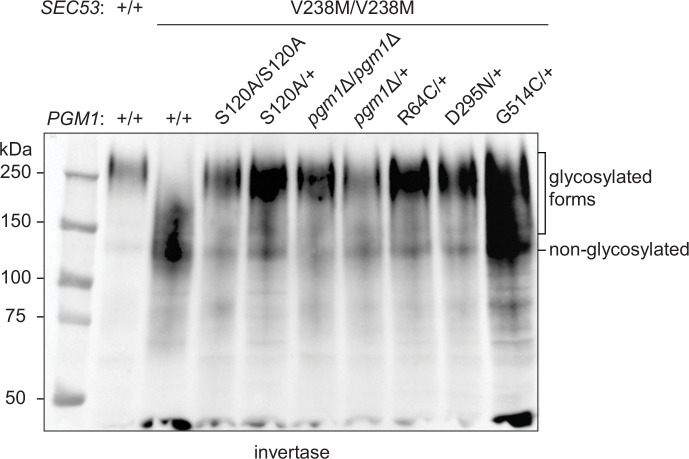
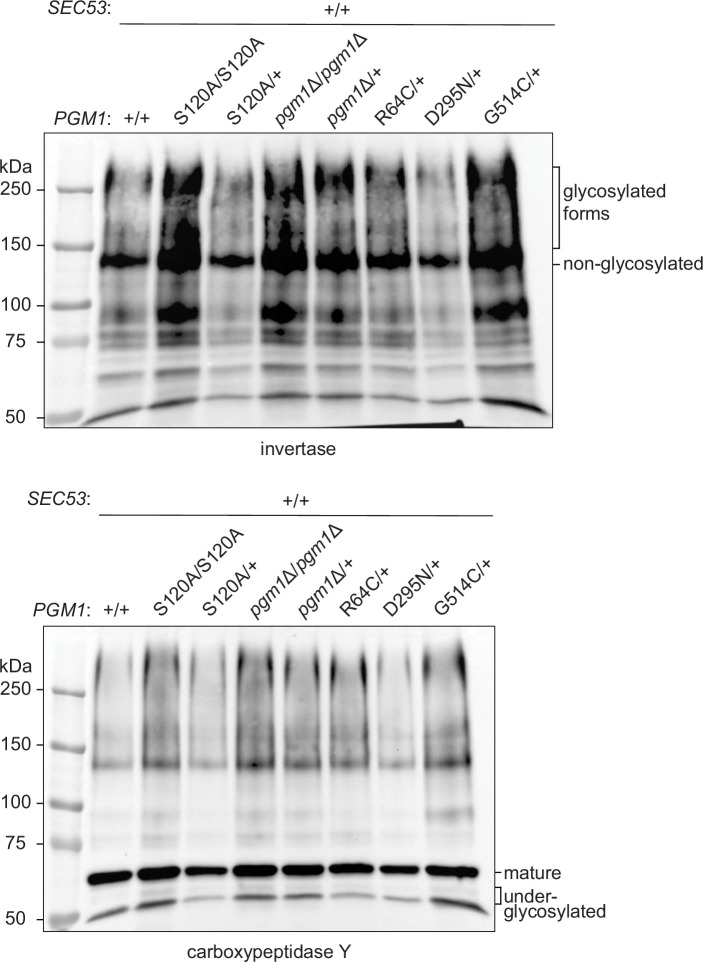
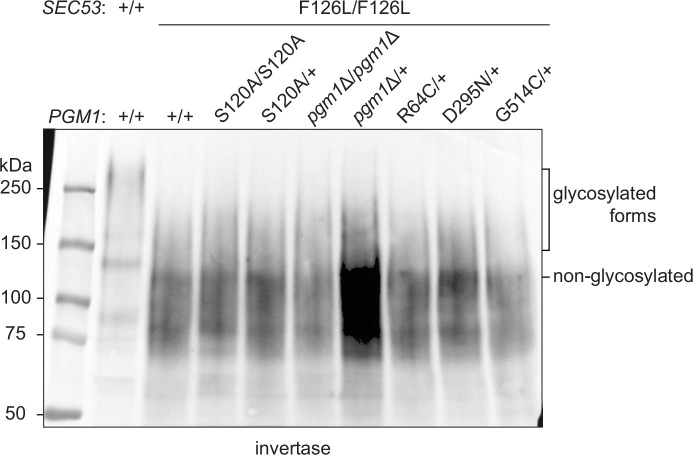
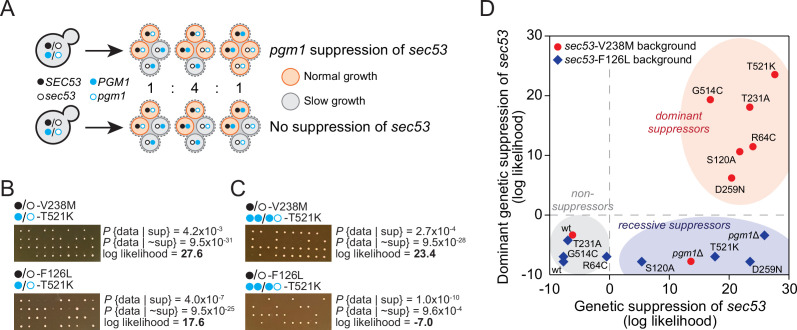
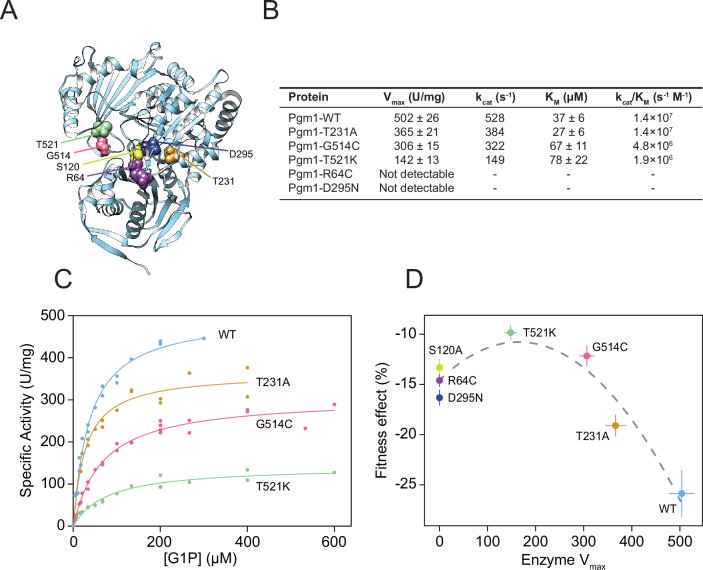
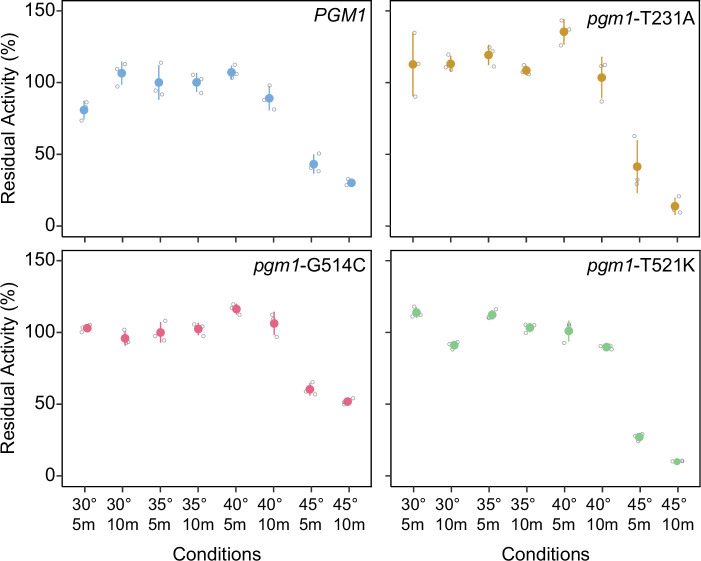
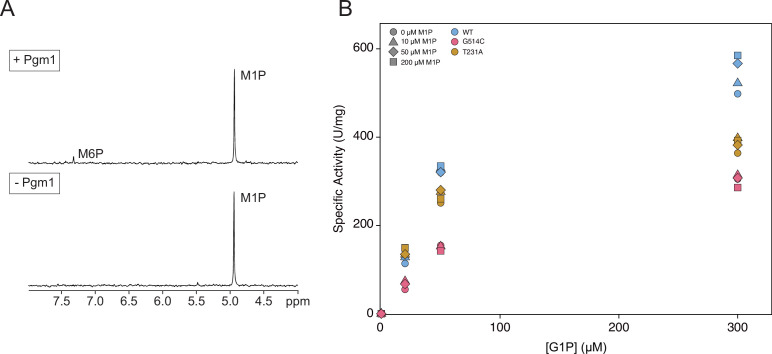
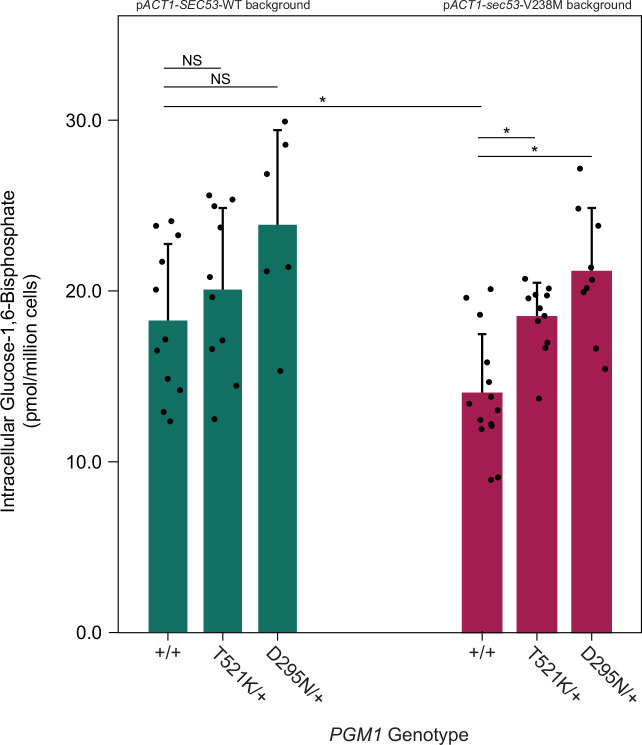
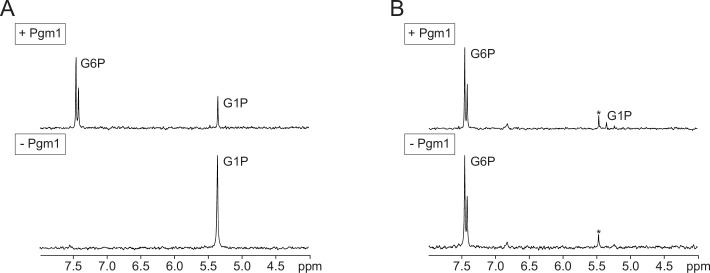
The most common cause of human congenital disorders of glycosylation (CDG) are mutations in the phosphomannomutase gene PMM2, which affect protein N-linked glycosylation. The yeast gene SEC53 encodes a homolog of human PMM2. We evolved 384 populations of yeast harboring one of two human-disease-associated alleles, sec53-V238M and sec53-F126L, or wild-type SEC53. We find that after 1000 generations, most populations compensate for the slow-growth phenotype associated with the sec53 human-disease-associated alleles. Through whole-genome sequencing we identify compensatory mutations, including known SEC53 genetic interactors. We observe an enrichment of compensatory mutations in other genes whose human homologs are associated with Type 1 CDG, including PGM1, which encodes the minor isoform of phosphoglucomutase in yeast. By genetic reconstruction, we show that evolved pgm1 mutations are dominant and allele-specific genetic interactors that restore both protein glycosylation and growth of yeast harboring the sec53-V238M allele. Finally, we characterize the enzymatic activity of purified Pgm1 mutant proteins. We find that reduction, but not elimination, of Pgm1 activity best compensates for the deleterious phenotypes associated with the sec53-V238M allele. Broadly, our results demonstrate the power of experimental evolution as a tool for identifying genes and pathways that compensate for human-disease-associated alleles.
Keywords: PMM2-CDG; S. cerevisiae; congenital disorders of glycosylation; evolutionary biology; experimental evolution; genetics; genomics; humanized yeast.
© 2022, Vignogna et al.
Conflict of interest statement
RV, MA, MM, JN, RS, EP, GA, GL No competing interests declared
Figures


References
-
- Aebi M, Helenius A, Schenk B, Barone R, Fiumara A, Berger EG, Hennet T, Imbach T, Stutz A, Bjursell C, Uller A, Wahlström JG, Briones P, Cardo E, Clayton P, Winchester B, Cormier-Dalre V, de Lonlay P, Cuer M, Dupré T, Seta N, de Koning T, Dorland L, de Loos F, Kupers L. Carbohydrate-deficient glycoprotein syndromes become congenital disorders of glycosylation: an updated nomenclature for CDG first international workshop on cdgs. Glycoconjugate Journal. 1999;16:669–671. doi: 10.1023/a:1017249723165. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
