

Linking nutrient sensing, mitochondrial function, and PRR immune cell signaling in liver disease
- PMID: 36216719
- PMCID: PMC9617785
- DOI: 10.1016/j.it.2022.09.002
Linking nutrient sensing, mitochondrial function, and PRR immune cell signaling in liver disease
Abstract
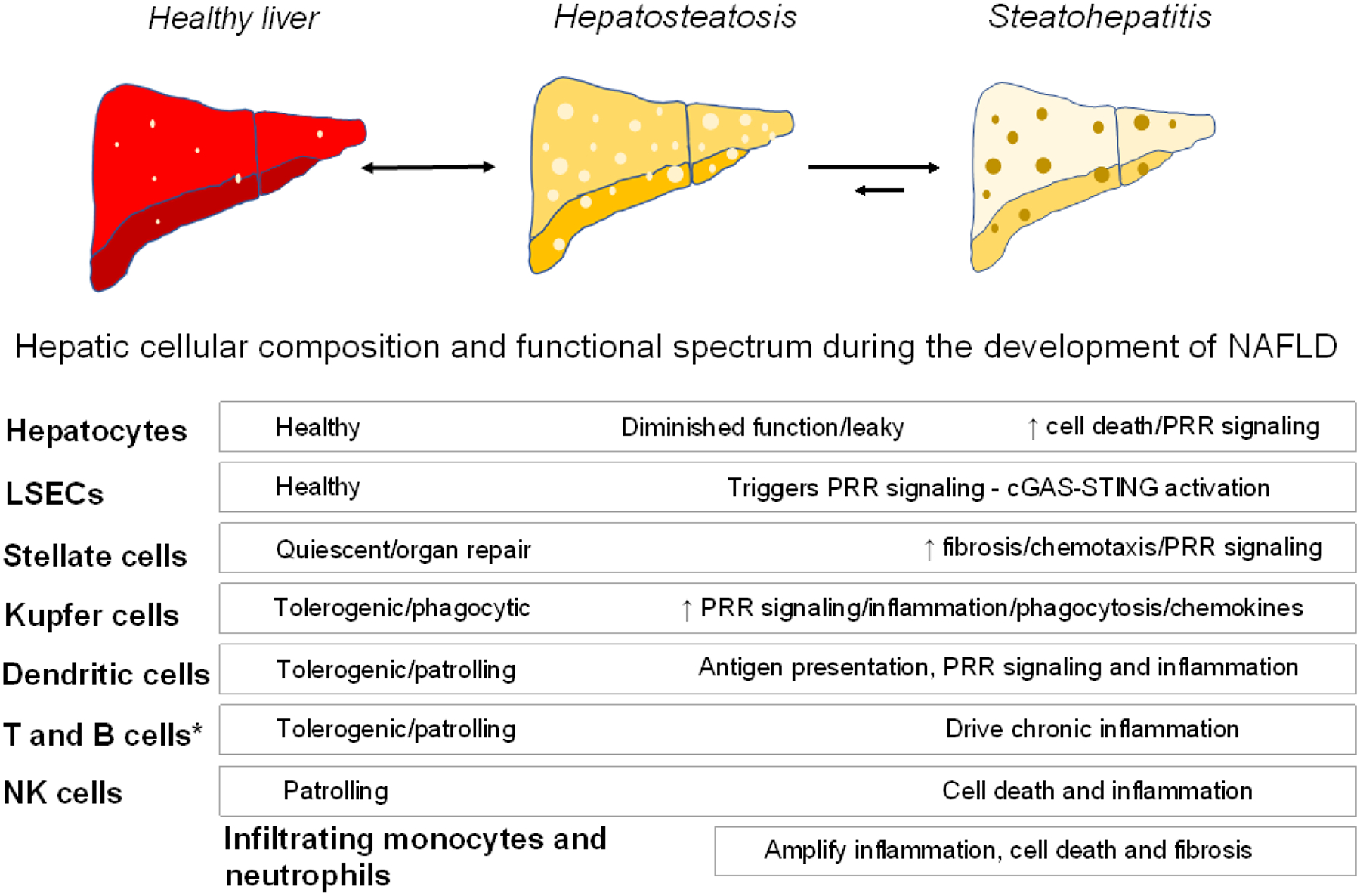
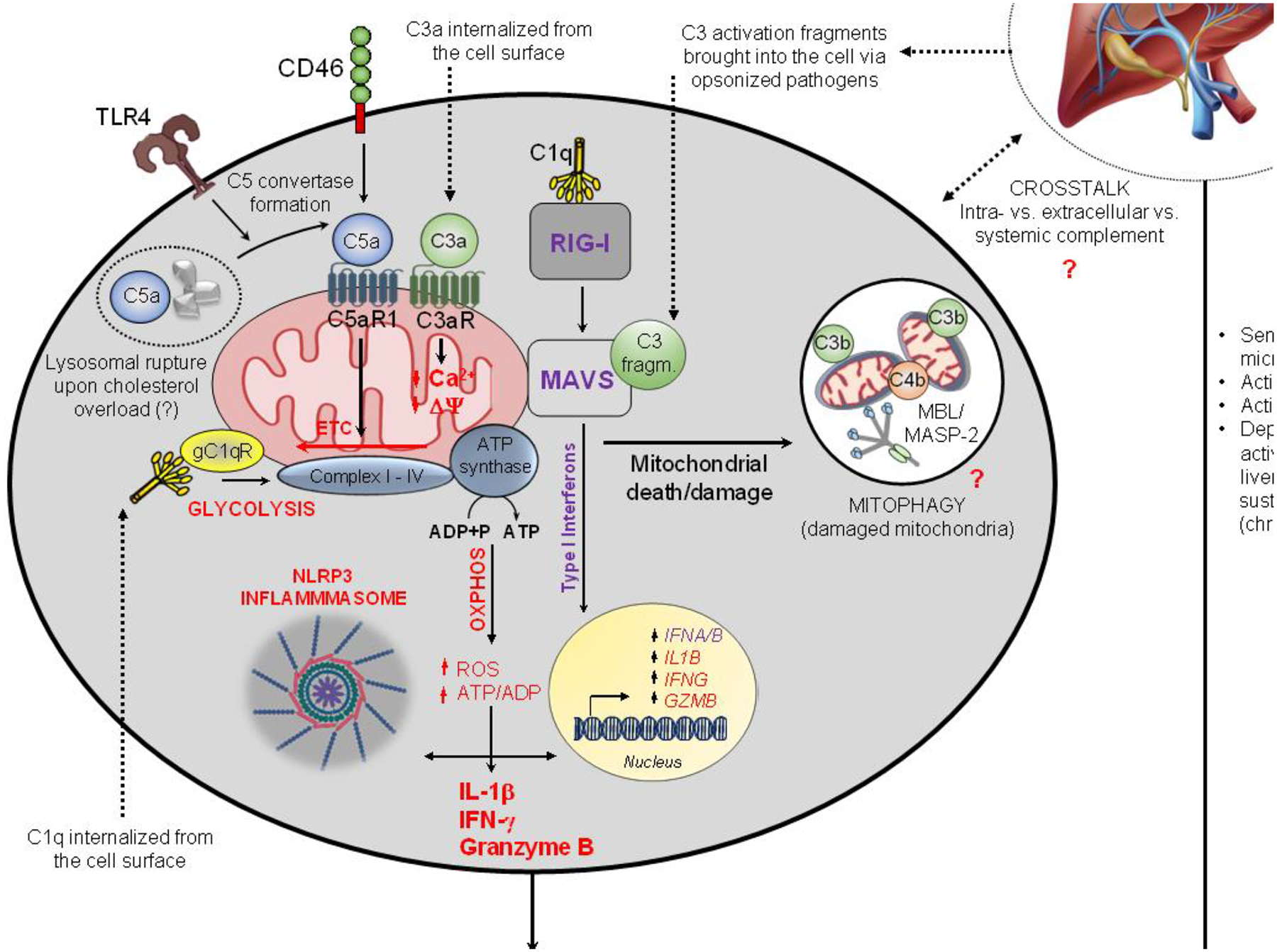
Caloric overconsumption in vertebrates promotes adipose and liver fat accumulation while perturbing the gut microbiome. This triad triggers pattern recognition receptor (PRR)-mediated immune cell signaling and sterile inflammation. Moreover, immune system activation perpetuates metabolic consequences, including the progression of nonalcoholic fatty liver disease (NAFLD) to nonalcoholic hepatic steatohepatitis (NASH). Recent findings show that sensing of nutrient overabundance disrupts the activity and homeostasis of the central cellular energy-generating organelle, the mitochondrion. In parallel, whether caloric excess-initiated PRR signaling and mitochondrial perturbations are coordinated to amplify this inflammatory process in NASH progression remains in question. We hypothesize that altered mitochondrial function, classic PRR signaling, and complement activation in response to nutrient overload together play an integrated role across the immune cell landscape, leading to liver inflammation and NASH progression.
Keywords: cardiometabolic disease; complement; liver; mitochondria; pattern recognition receptors.
Published by Elsevier Ltd.
Conflict of interest statement
Declaration of interests No interests are declared.
Figures


References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
