

Drugging p53 in cancer: one protein, many targets
- PMID: 36216888
- PMCID: PMC9549847
- DOI: 10.1038/s41573-022-00571-8
Drugging p53 in cancer: one protein, many targets
Abstract
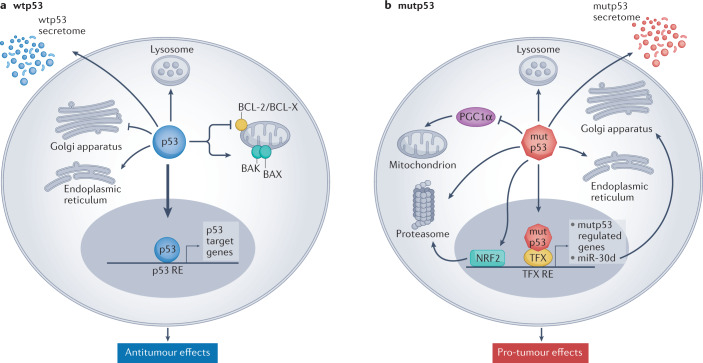
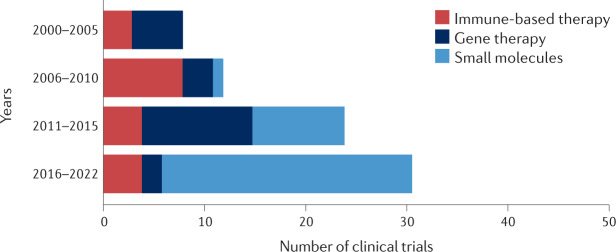
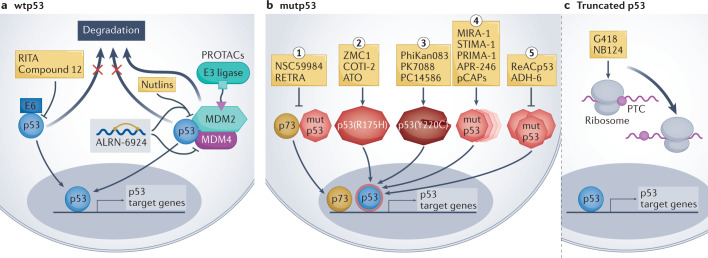
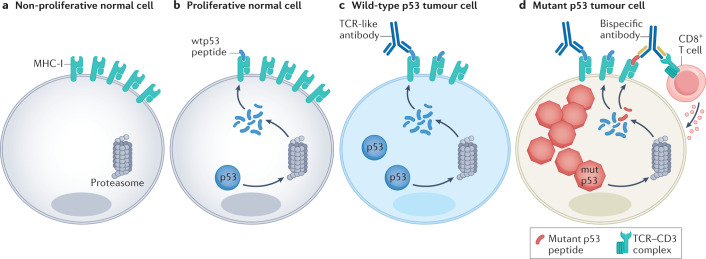
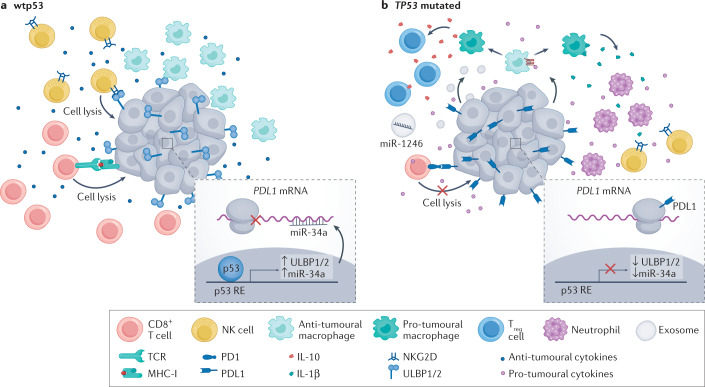
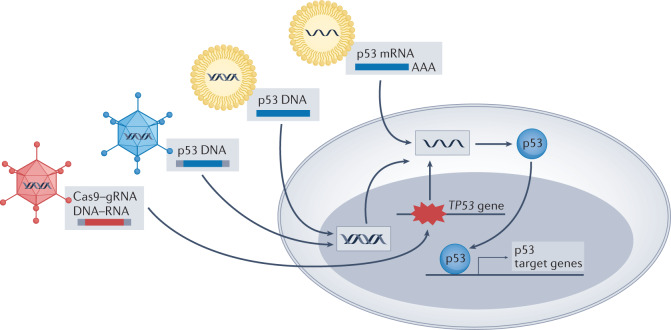
Mutations in the TP53 tumour suppressor gene are very frequent in cancer, and attempts to restore the functionality of p53 in tumours as a therapeutic strategy began decades ago. However, very few of these drug development programmes have reached late-stage clinical trials, and no p53-based therapeutics have been approved in the USA or Europe so far. This is probably because, as a nuclear transcription factor, p53 does not possess typical drug target features and has therefore long been considered undruggable. Nevertheless, several promising approaches towards p53-based therapy have emerged in recent years, including improved versions of earlier strategies and novel approaches to make undruggable targets druggable. Small molecules that can either protect p53 from its negative regulators or restore the functionality of mutant p53 proteins are gaining interest, and drugs tailored to specific types of p53 mutants are emerging. In parallel, there is renewed interest in gene therapy strategies and p53-based immunotherapy approaches. However, major concerns still remain to be addressed. This Review re-evaluates the efforts made towards targeting p53-dysfunctional cancers, and discusses the challenges encountered during clinical development.
© 2022. Springer Nature Limited.
Conflict of interest statement
M.O. consults for Quintrigen.
Figures
References
-
- Bykov VJN, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer. 2017;182:89–102. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
