

Pneumococcal within-host diversity during colonization, transmission and treatment
- PMID: 36216891
- PMCID: PMC9613479
- DOI: 10.1038/s41564-022-01238-1
Pneumococcal within-host diversity during colonization, transmission and treatment
Abstract
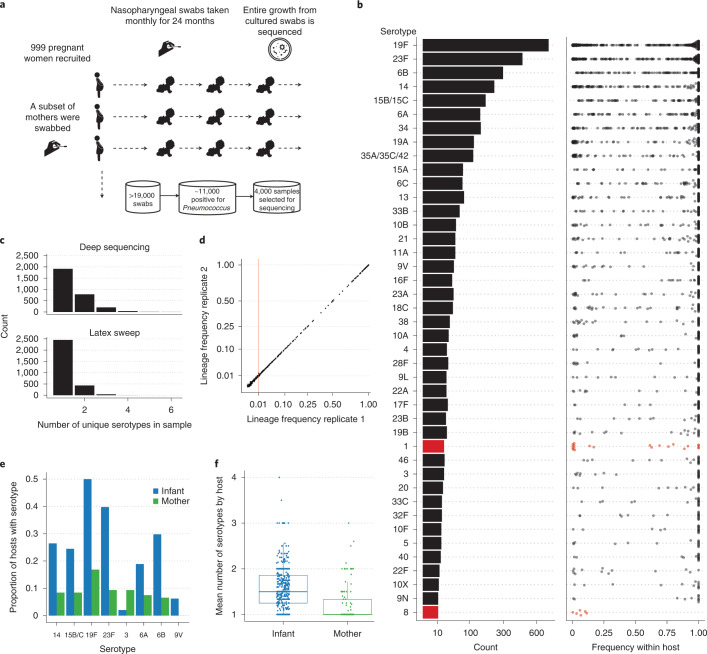
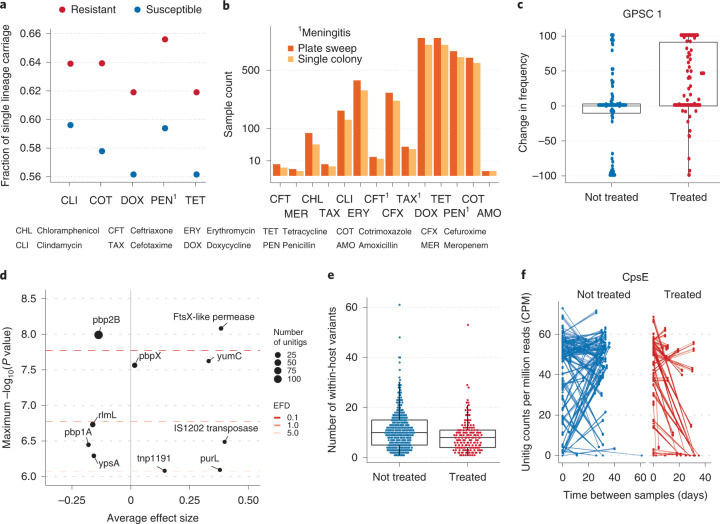
Characterizing the genetic diversity of pathogens within the host promises to greatly improve surveillance and reconstruction of transmission chains. For bacteria, it also informs our understanding of inter-strain competition and how this shapes the distribution of resistant and sensitive bacteria. Here we study the genetic diversity of Streptococcus pneumoniae within 468 infants and 145 of their mothers by deep sequencing whole pneumococcal populations from 3,761 longitudinal nasopharyngeal samples. We demonstrate that deep sequencing has unsurpassed sensitivity for detecting multiple colonization, doubling the rate at which highly invasive serotype 1 bacteria were detected in carriage compared with gold-standard methods. The greater resolution identified an elevated rate of transmission from mothers to their children in the first year of the child's life. Comprehensive treatment data demonstrated that infants were at an elevated risk of both the acquisition and persistent colonization of a multidrug-resistant bacterium following antimicrobial treatment. Some alleles were enriched after antimicrobial treatment, suggesting that they aided persistence, but generally purifying selection dominated within-host evolution. Rates of co-colonization imply that in the absence of treatment, susceptible lineages outcompeted resistant lineages within the host. These results demonstrate the many benefits of deep sequencing for the genomic surveillance of bacterial pathogens.
© 2022. The Author(s).
Conflict of interest statement
N.J.C. was a consultant for Antigen Discovery, Inc., involved in the design of a proteome array for
Figures












References
-
- GBD 2016 Lower Respiratory Infections Collaborators. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory infections in 195 countries, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect. Dis. 2018;18:1191–1210. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
