

Dynamic SARS-CoV-2 emergence algorithm for rationally-designed logical next-generation vaccines
- PMID: 36217024
- PMCID: PMC9550860
- DOI: 10.1038/s42003-022-04030-3
Dynamic SARS-CoV-2 emergence algorithm for rationally-designed logical next-generation vaccines
Abstract
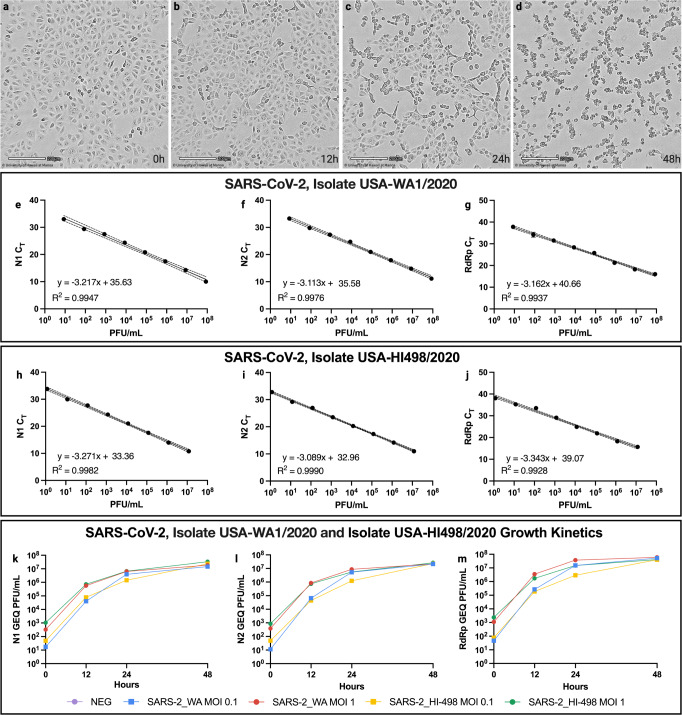
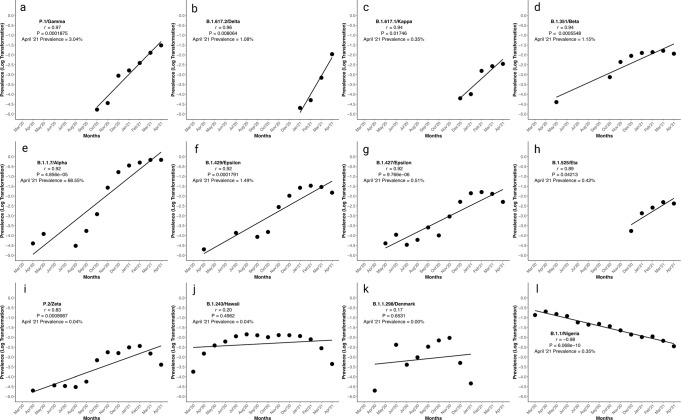
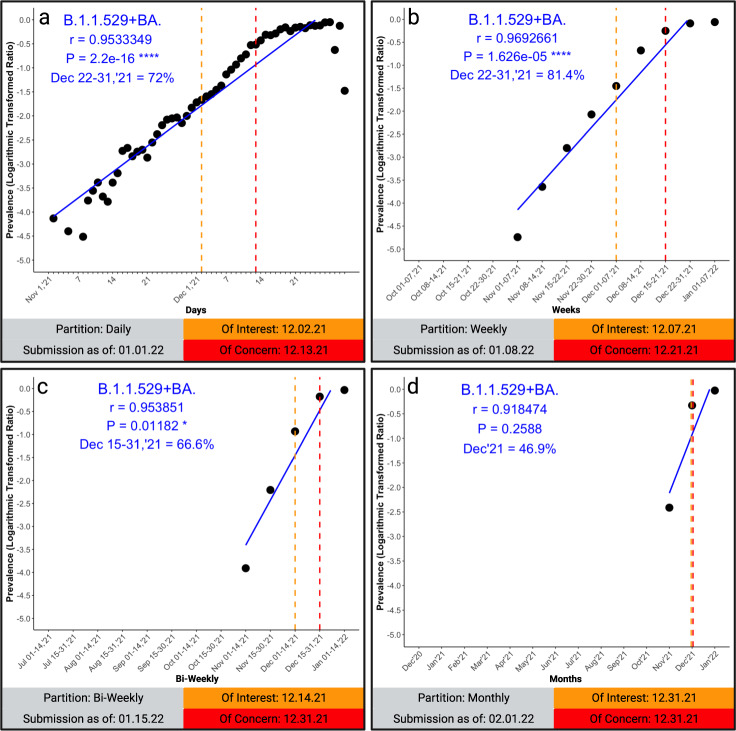
SARS-CoV-2 worldwide spread and evolution has resulted in variants containing mutations resulting in immune evasive epitopes that decrease vaccine efficacy. We acquired SARS-CoV-2 positive clinical samples and compared the worldwide emerged spike mutations from Variants of Concern/Interest, and developed an algorithm for monitoring the evolution of SARS-CoV-2 in the context of vaccines and monoclonal antibodies. The algorithm partitions logarithmic-transformed prevalence data monthly and Pearson's correlation determines exponential emergence of amino acid substitutions (AAS) and lineages. The SARS-CoV-2 genome evaluation indicated 49 mutations, with 44 resulting in AAS. Nine of the ten most worldwide prevalent (>70%) spike protein changes have Pearson's coefficient r > 0.9. The tenth, D614G, has a prevalence >99% and r-value of 0.67. The resulting algorithm is based on the patterns these ten substitutions elucidated. The strong positive correlation of the emerged spike protein changes and algorithmic predictive value can be harnessed in designing vaccines with relevant immunogenic epitopes. Monitoring, next-generation vaccine design, and mAb clinical efficacy must keep up with SARS-CoV-2 evolution, as the virus is predicted to remain endemic.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


References
-
- CDC. Cases, Data, and Surveillance. Centers for Disease Control and Prevention. 2020. https://www.cdc.gov/coronavirus/2019-ncov/cases-updates/variant-surveill...
-
- Tracking SARS-CoV-2 variants. World Health Organization. Published July 6, 2021. Accessed July 16, 2021. https://www.who.int/activities/tracking-SARS-CoV-2-variants
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
