

A proteome-scale map of the SARS-CoV-2-human contactome
- PMID: 36217029
- PMCID: PMC9849141
- DOI: 10.1038/s41587-022-01475-z
A proteome-scale map of the SARS-CoV-2-human contactome
Abstract
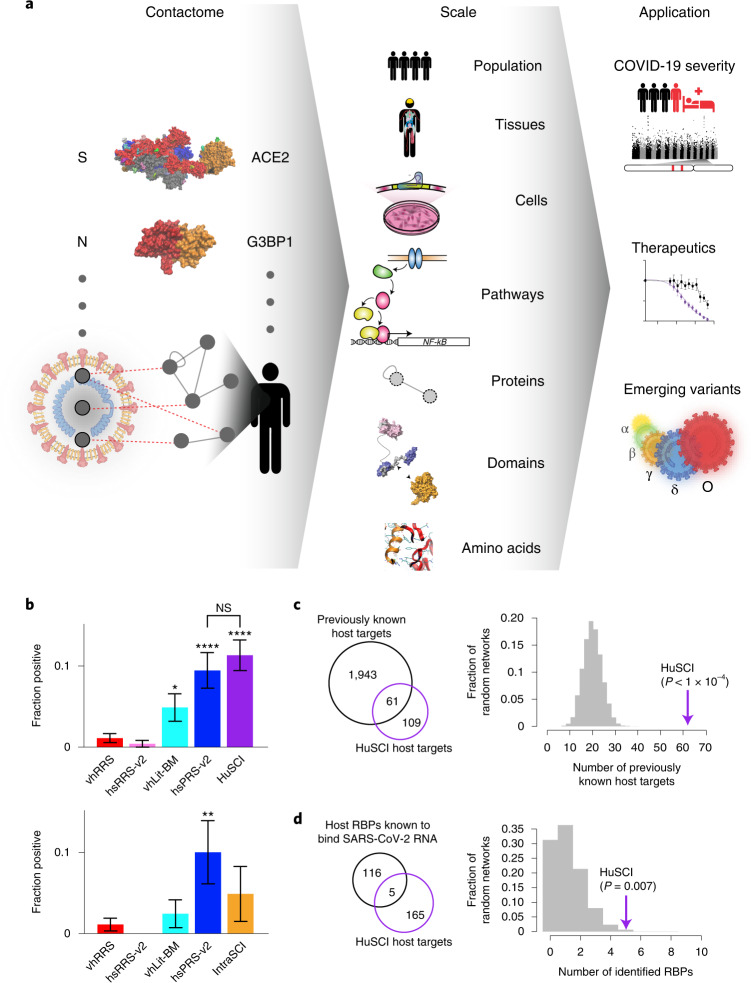
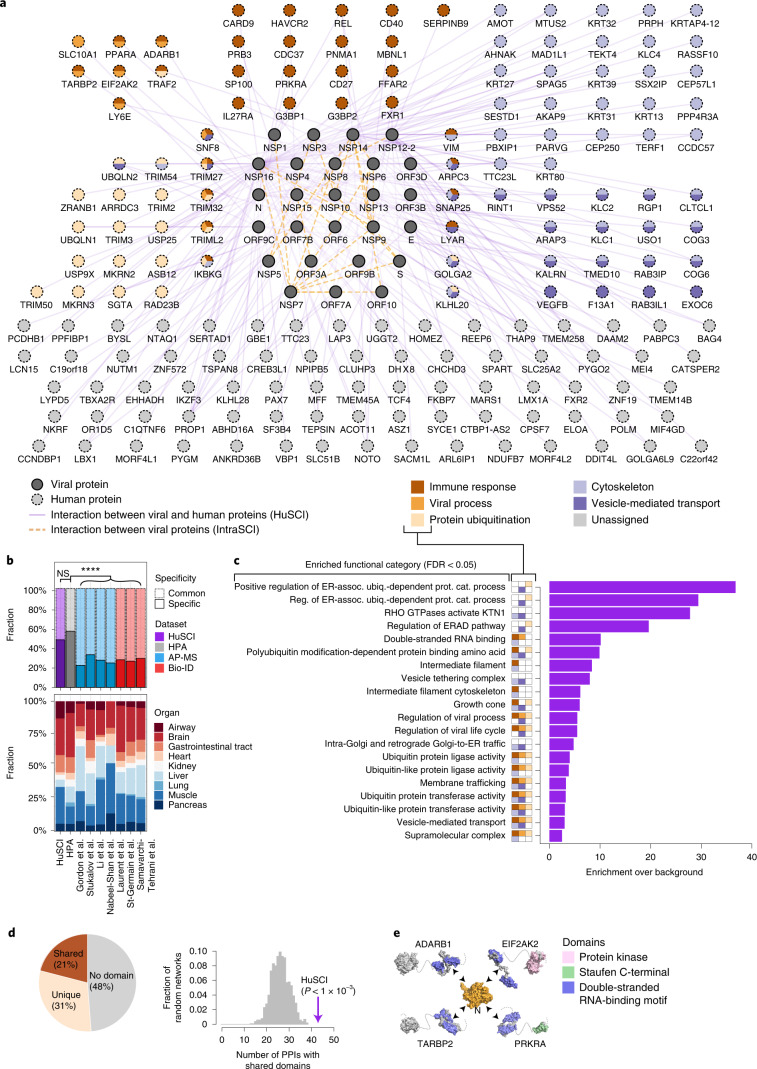
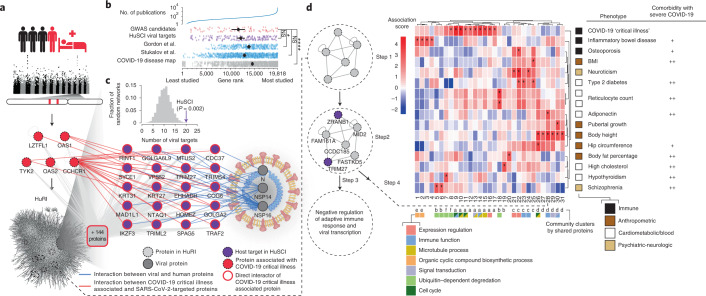
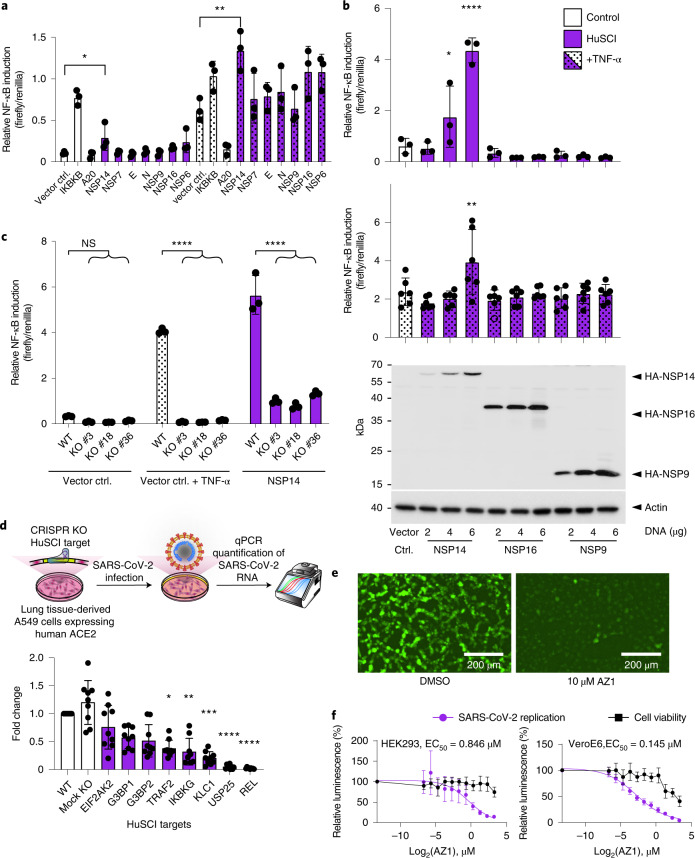
Understanding the mechanisms of coronavirus disease 2019 (COVID-19) disease severity to efficiently design therapies for emerging virus variants remains an urgent challenge of the ongoing pandemic. Infection and immune reactions are mediated by direct contacts between viral molecules and the host proteome, and the vast majority of these virus-host contacts (the 'contactome') have not been identified. Here, we present a systematic contactome map of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) with the human host encompassing more than 200 binary virus-host and intraviral protein-protein interactions. We find that host proteins genetically associated with comorbidities of severe illness and long COVID are enriched in SARS-CoV-2 targeted network communities. Evaluating contactome-derived hypotheses, we demonstrate that viral NSP14 activates nuclear factor κB (NF-κB)-dependent transcription, even in the presence of cytokine signaling. Moreover, for several tested host proteins, genetic knock-down substantially reduces viral replication. Additionally, we show for USP25 that this effect is phenocopied by the small-molecule inhibitor AZ1. Our results connect viral proteins to human genetic architecture for COVID-19 severity and offer potential therapeutic targets.
© 2022. The Author(s).
Conflict of interest statement
F.R. and M.V. are advisors and shareholders of SeqWell, Inc. (Beverly, MA, USA).
Figures
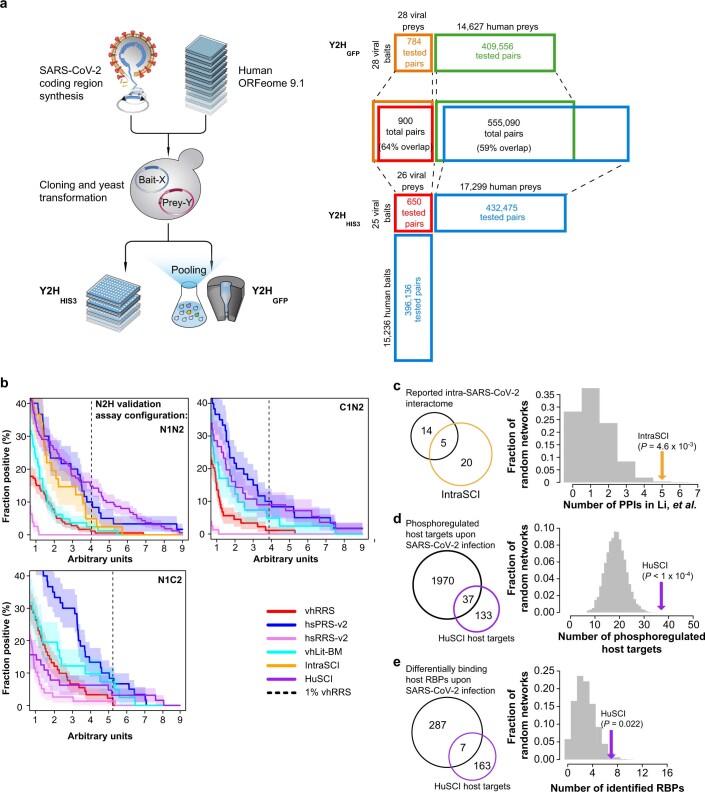
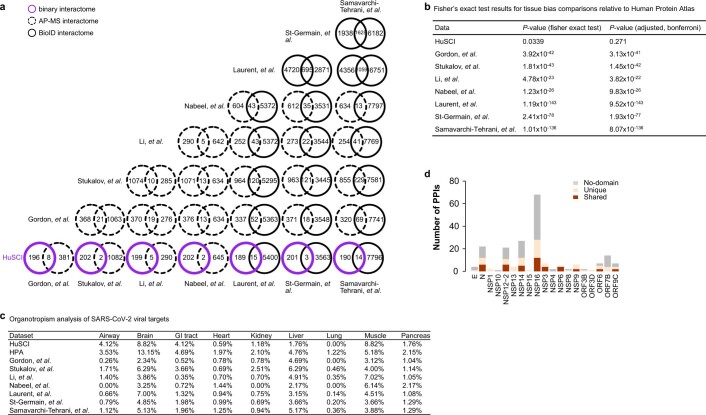

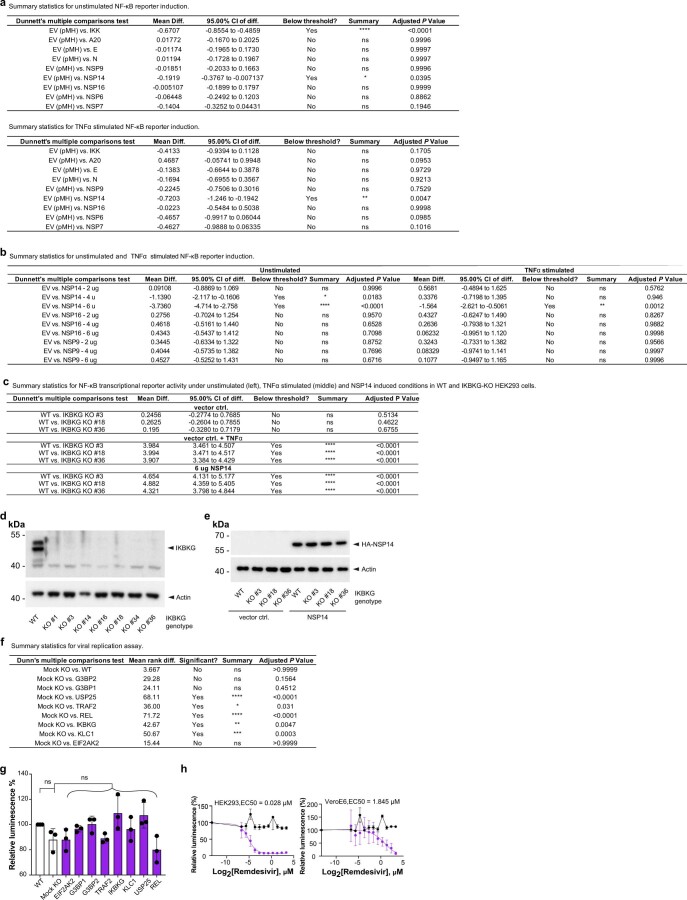
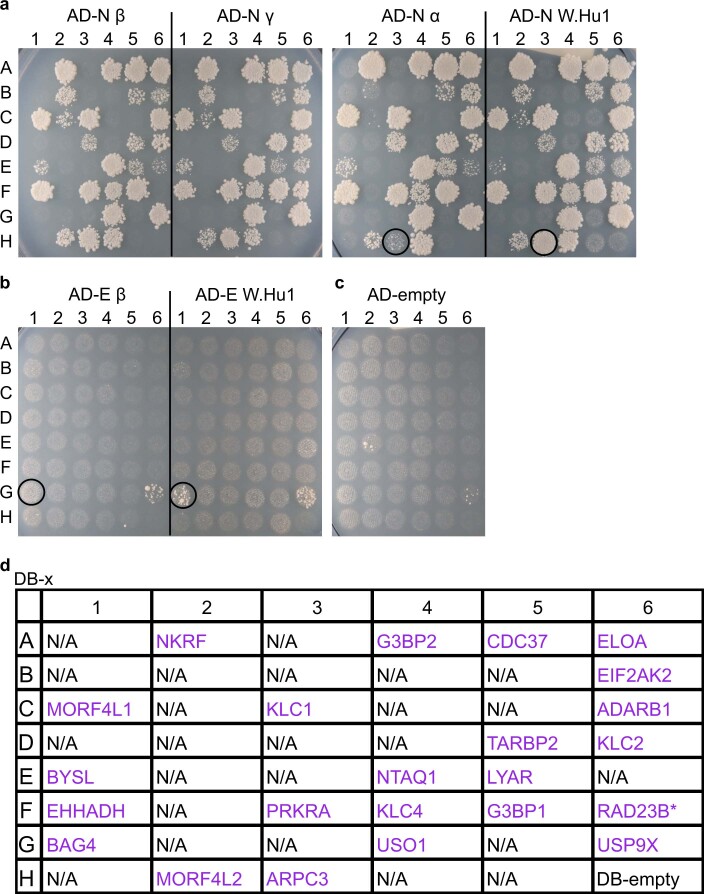
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
