

Comparative Genomic Analysis of Fusobacterium necrophorum Provides Insights into Conserved Virulence Genes
- PMID: 36219094
- PMCID: PMC9769765
- DOI: 10.1128/spectrum.00297-22
Comparative Genomic Analysis of Fusobacterium necrophorum Provides Insights into Conserved Virulence Genes
Abstract
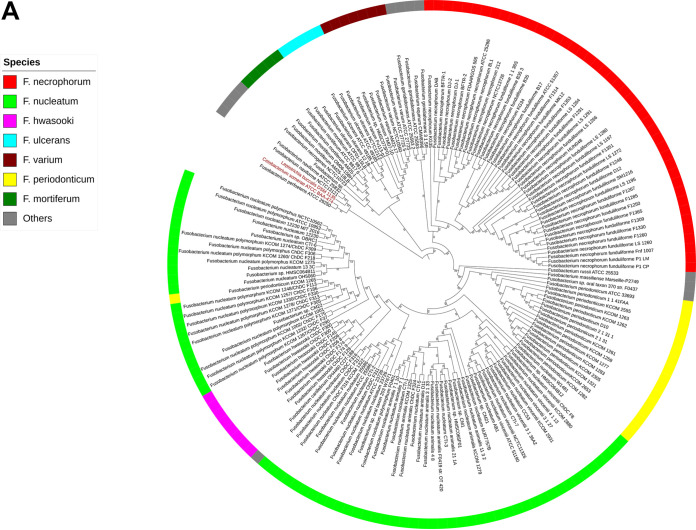
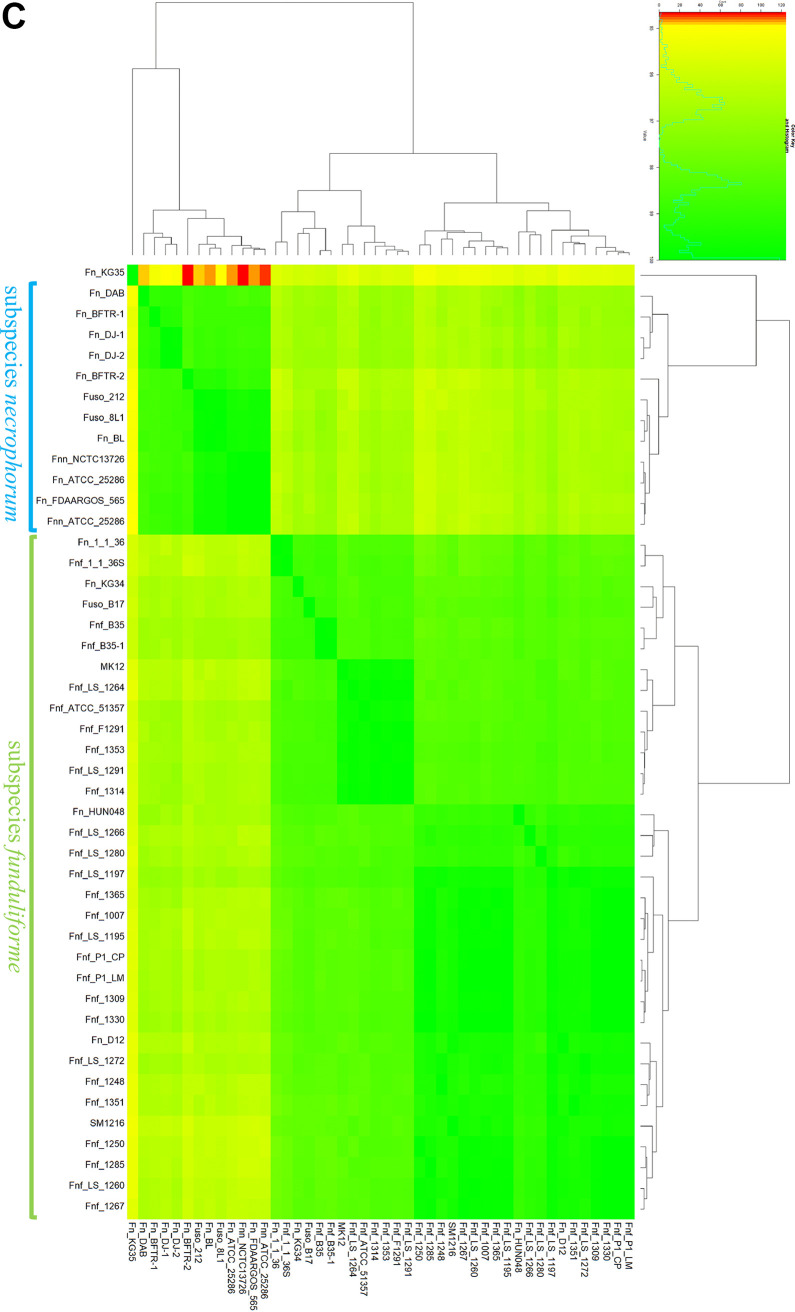
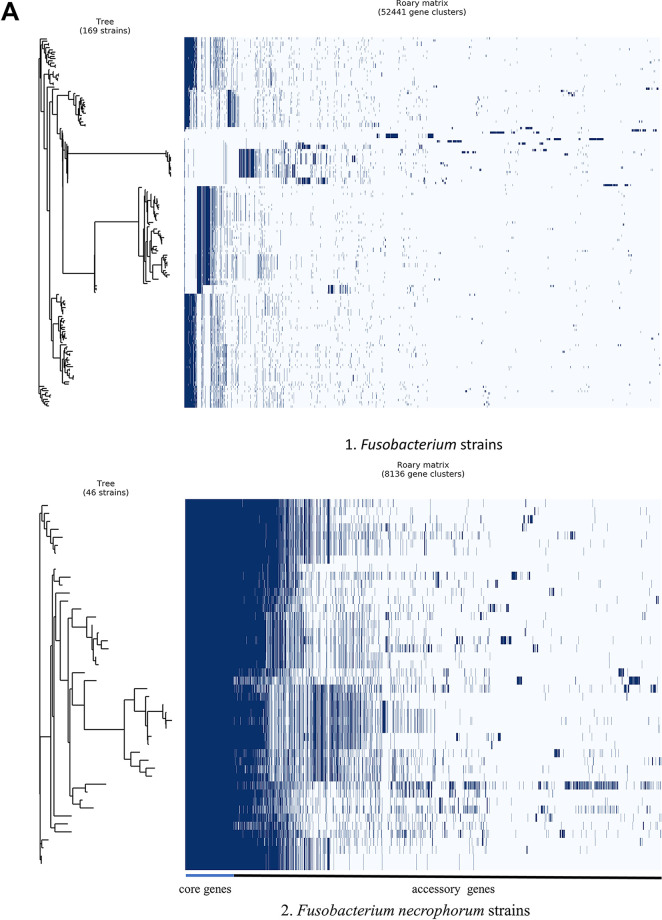
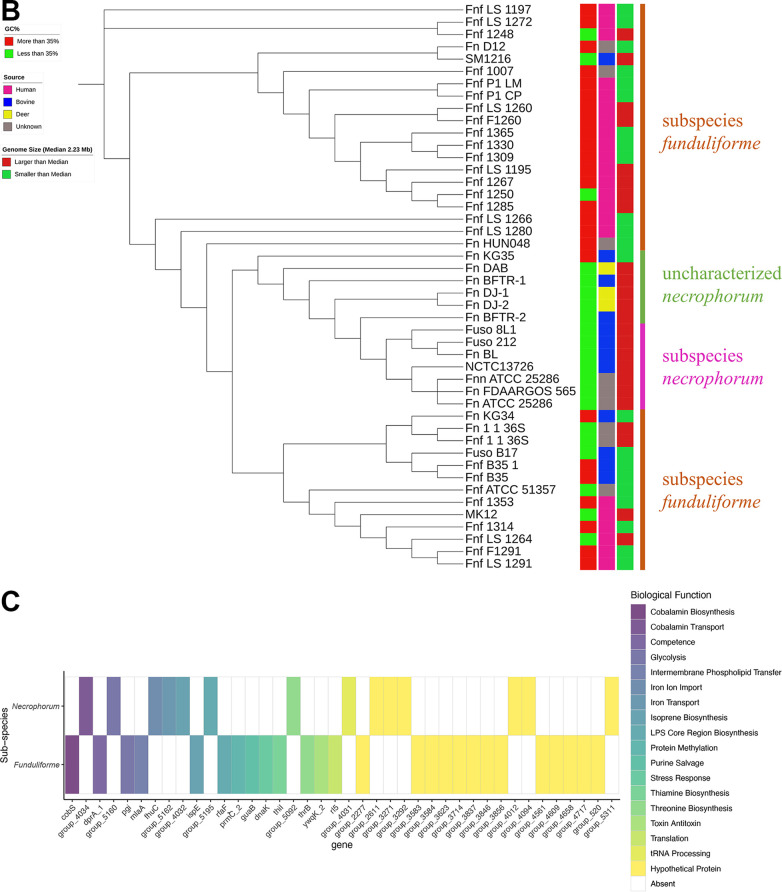
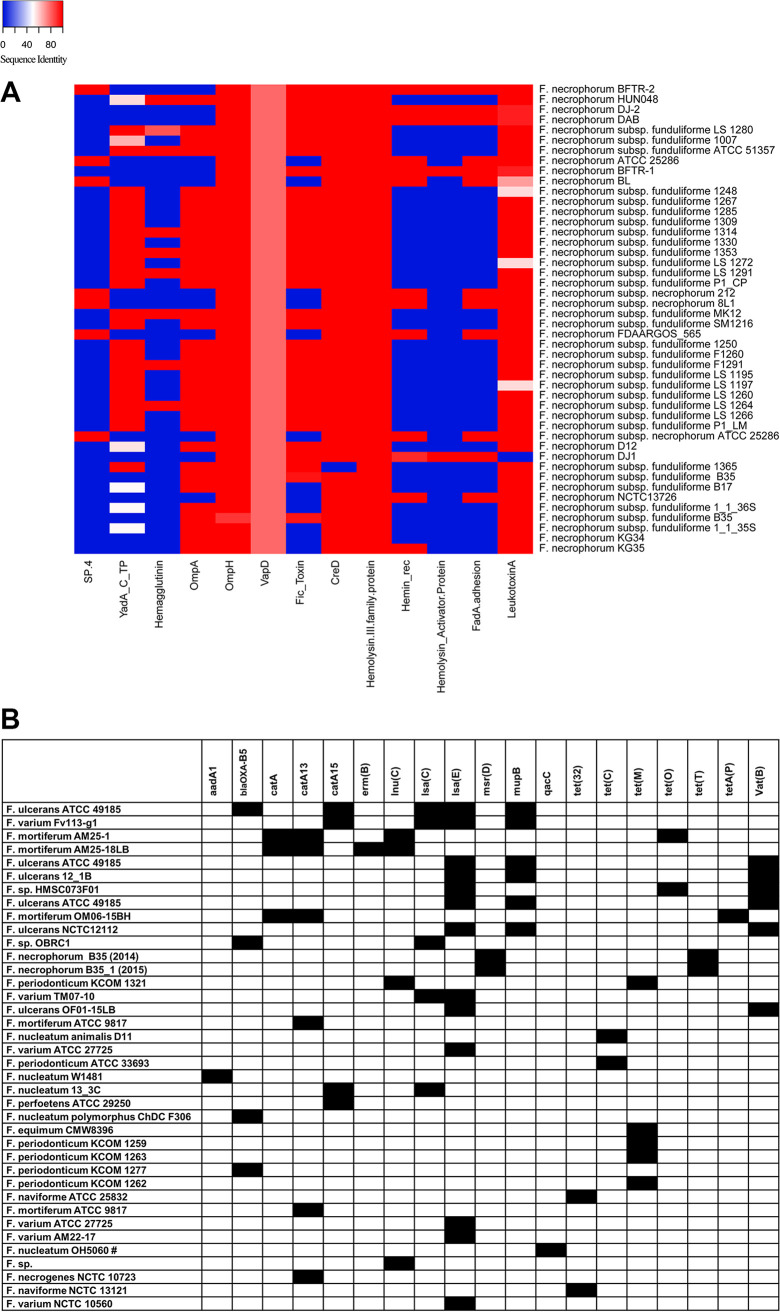
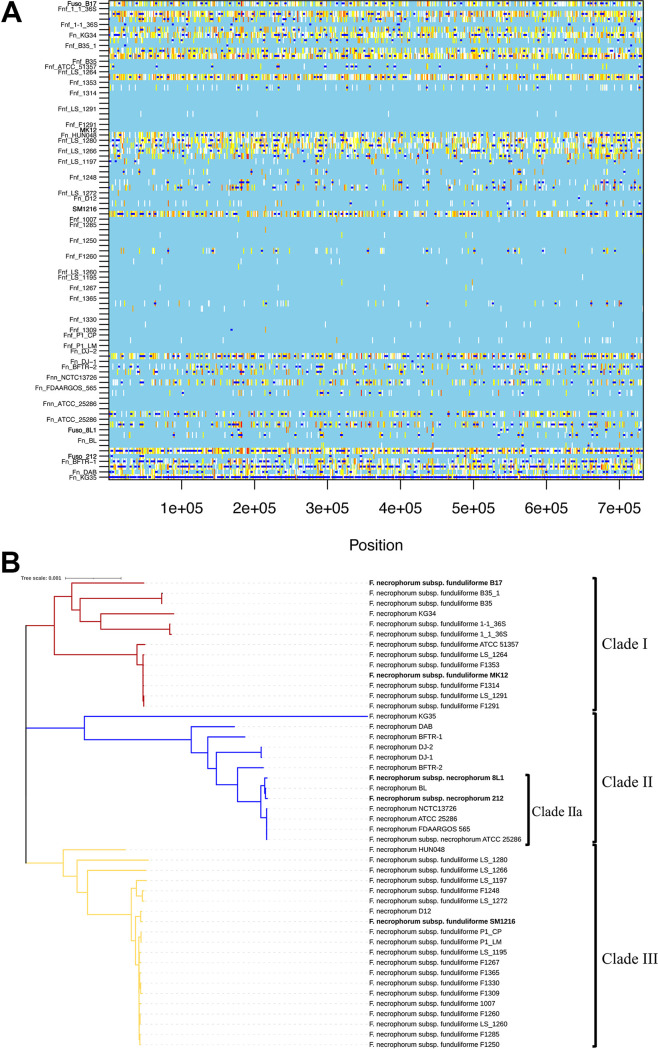
Fusobacterium necrophorum is a Gram-negative, filamentous anaerobe prevalent in the mucosal flora of animals and humans. It causes necrotic infections in cattle, resulting in a substantial economic impact on the cattle industry. Although infection severity and management differ within F. necrophorum species, little is known about F. necrophorum speciation and the genetic virulence determinants between strains. To characterize the clinical isolates, we performed whole-genome sequencing of four bovine isolates (8L1, 212, B17, and SM1216) and one human isolate (MK12). To determine the phylogenetic relationship and evolution pattern and investigate the presence of antimicrobial resistance genes (ARGs) and potential virulence genes of F. necrophorum, we also performed comparative genomics with publicly available Fusobacterium genomes. Using up-to-date bacterial core gene (UBCG) set analysis, we uncovered distinct Fusobacterium species and F. necrophorum subspecies clades. Pangenome analyses revealed a high level of diversity among Fusobacterium strains down to species levels. The output also identified 14 and 26 genes specific to F. necrophorum subsp. necrophorum and F. necrophorum subsp. funduliforme, respectively, which could be essential for bacterial survival under different environmental conditions. ClonalFrameML-based recombination analysis suggested that extensive recombination among accessory genes led to species divergence. Furthermore, the only strain of F. necrophorum with ARGs was F. necrophorum subsp. funduliforme B35, with acquired macrolide and tetracycline resistance genes. Our custom search revealed common virulence genes, including toxins, adhesion proteins, outer membrane proteins, cell envelope, type IV secretion system, ABC (ATP-binding cassette) transporters, and transporter proteins. A focused study on these genes could help identify major virulence genes and inform effective vaccination strategies against fusobacterial infections. IMPORTANCE Fusobacterium necrophorum is an anaerobic bacterium that causes liver abscesses in cattle with an annual incidence rate of 10% to 20%, resulting in a substantial economic impact on the cattle industry. The lack of definite biochemical tests makes it difficult to distinguish F. necrophorum subspecies phenotypically, where genomic characterization plays a significant role. However, due to the lack of a good reference genome for comparison, F. necrophorum subspecies-level identification represents a significant challenge. To overcome this challenge, we used comparative genomics to validate clinical test strains for subspecies-level identification. The findings of our study help predict specific clades of previously uncharacterized strains of F. necrophorum. Our study identifies both general and subspecies-specific virulence genes through a custom search-based analysis. The virulence genes identified in this study can be the focus of future studies aimed at evaluating their potential as vaccine targets to prevent fusobacterial infections in cattle.
Keywords: Fusobacterium necrophorum; pangenome; phylogeny; recombination; subspecies; virulence factors; virulence genes.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Human Fusobacterium necrophorum strains have a leukotoxin gene and exhibit leukotoxic activity.J Med Microbiol. 2008 Feb;57(Pt 2):225-231. doi: 10.1099/jmm.0.47598-0. J Med Microbiol. 2008. PMID: 18201990
-
Ribotyping to differentiate Fusobacterium necrophorum subsp. necrophorum and F. necrophorum subsp. funduliforme isolated from bovine ruminal contents and liver abscesses.Appl Environ Microbiol. 1996 Feb;62(2):469-72. doi: 10.1128/aem.62.2.469-472.1996. Appl Environ Microbiol. 1996. PMID: 8593050 Free PMC article.
-
Identification of an outer membrane protein of Fusobacterium necrophorum subsp. necrophorum that binds with high affinity to bovine endothelial cells.Vet Microbiol. 2015 Mar 23;176(1-2):196-201. doi: 10.1016/j.vetmic.2014.12.015. Epub 2014 Dec 24. Vet Microbiol. 2015. PMID: 25601800
-
Fusobacterium necrophorum infections: virulence factors, pathogenic mechanism and control measures.Vet Res Commun. 1996;20(2):113-40. doi: 10.1007/BF00385634. Vet Res Commun. 1996. PMID: 8711893 Review.
-
Three variants of the leukotoxin gene in human isolates of Fusobacterium necrophorum subspecies funduliforme.Anaerobe. 2017 Jun;45:129-132. doi: 10.1016/j.anaerobe.2017.03.016. Epub 2017 Mar 19. Anaerobe. 2017. PMID: 28330774 Review.
Cited by
-
Association of pathogenic determinants of Fusobacterium necrophorum with bacteremia, and Lemierre's syndrome.Sci Rep. 2024 Aug 27;14(1):19804. doi: 10.1038/s41598-024-70608-y. Sci Rep. 2024. PMID: 39191804 Free PMC article.
-
A Global Comparative Genomic Analysis of Major Bacterial Pathogens in Bovine Mastitis and Lameness.Animals (Basel). 2025 Jan 30;15(3):394. doi: 10.3390/ani15030394. Animals (Basel). 2025. PMID: 39943164 Free PMC article. Review.
-
Characterization of Three New Outer Membrane Adhesion Proteins in Fusobacterium necrophorum.Microorganisms. 2023 Dec 12;11(12):2968. doi: 10.3390/microorganisms11122968. Microorganisms. 2023. PMID: 38138112 Free PMC article.
-
Unraveling the microbial diversity of bovine liver abscesses: isolation, identification, and genomic characterization of the Bacteroides found in hepatic lesions.Microbiol Spectr. 2025 Jun 3;13(6):e0042325. doi: 10.1128/spectrum.00423-25. Epub 2025 Apr 17. Microbiol Spectr. 2025. PMID: 40243342 Free PMC article.
-
Use of CRISPR interference for efficient and rapid gene inactivation in Fusobacterium nucleatum.Appl Environ Microbiol. 2024 Feb 21;90(2):e0166523. doi: 10.1128/aem.01665-23. Epub 2024 Jan 8. Appl Environ Microbiol. 2024. PMID: 38185820 Free PMC article.
References
-
- Finegold SM. 1996. Anaerobic Gram-negative bacilli, 4.11.1–4.11.17. In Baron S (ed), Medical Microbiology, 4th ed. University of Texas Medical Branch at Galveston, Galveston, TX. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
