

Acquired mutations in BAX confer resistance to BH3-mimetic therapy in acute myeloid leukemia
- PMID: 36219880
- PMCID: PMC10651776
- DOI: 10.1182/blood.2022016090
Acquired mutations in BAX confer resistance to BH3-mimetic therapy in acute myeloid leukemia
Erratum in
-
Moujalled DM, Brown FC, Chua CC, et al. Acquired mutations in BAX confer resistance to BH3-mimetic therapy in acute myeloid leukemia. Blood. 2023;141(6):634-644.Blood. 2023 Aug 3;142(5):494. doi: 10.1182/blood.2023020968. Blood. 2023. PMID: 37535365 Free PMC article. No abstract available.
Abstract
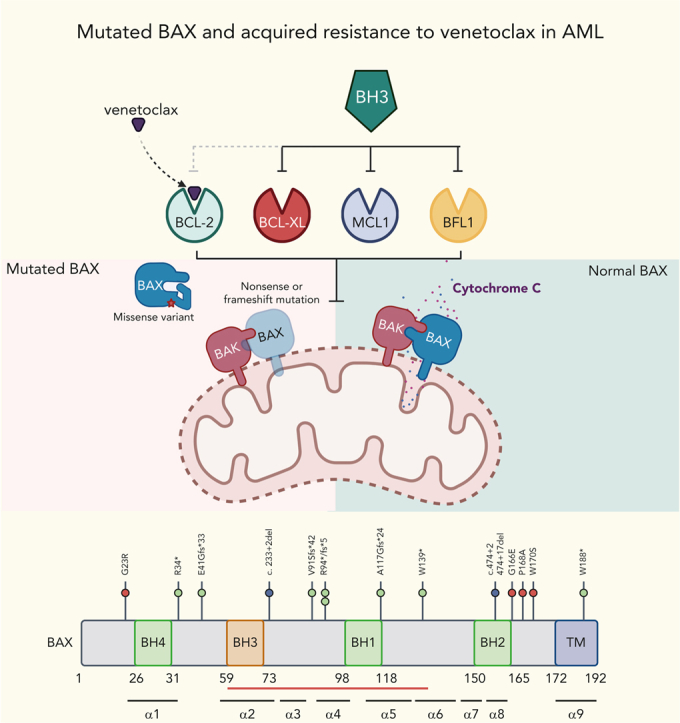
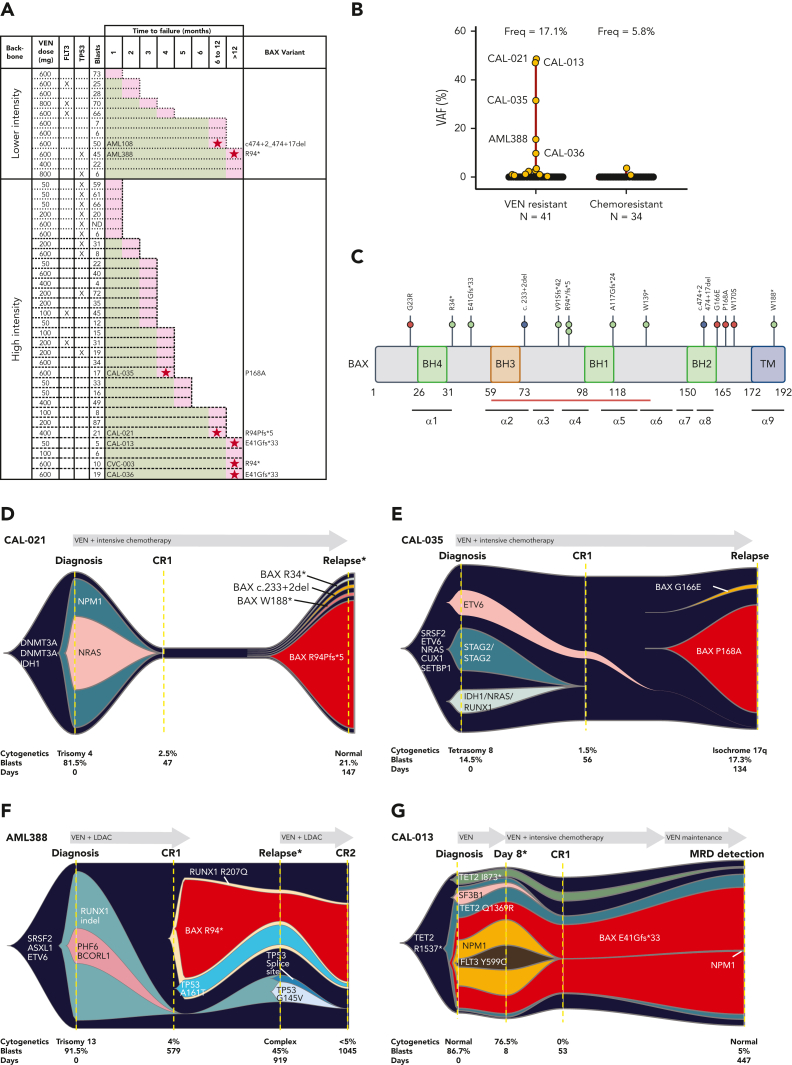
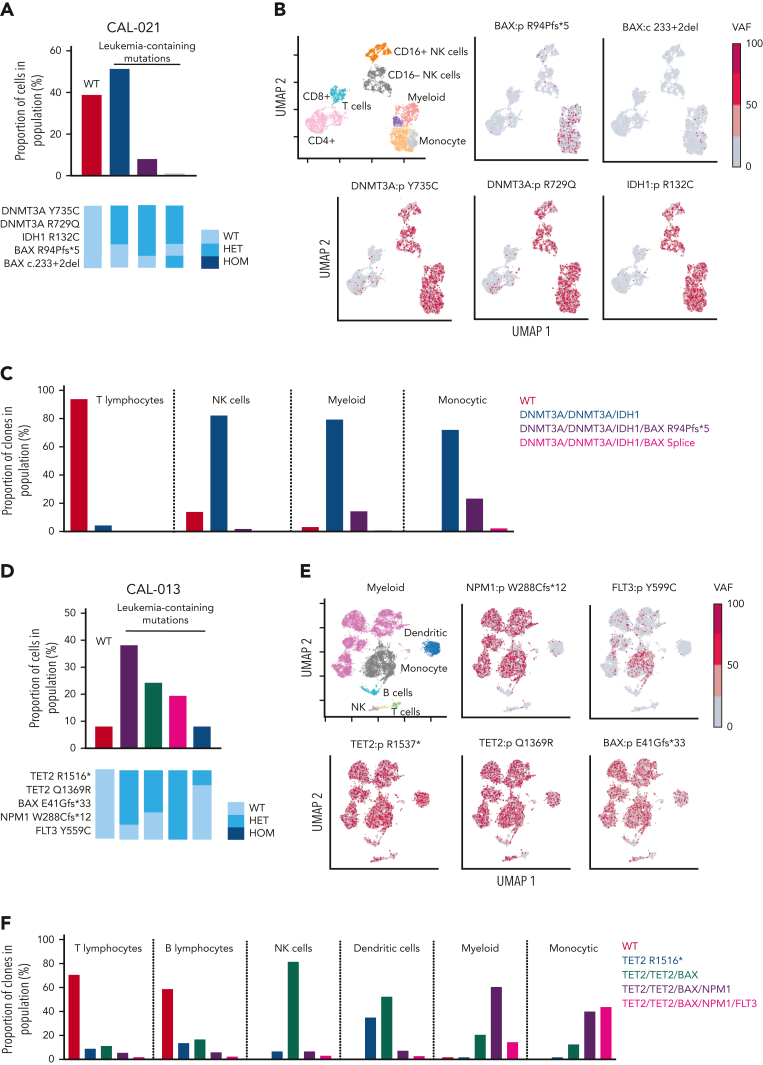
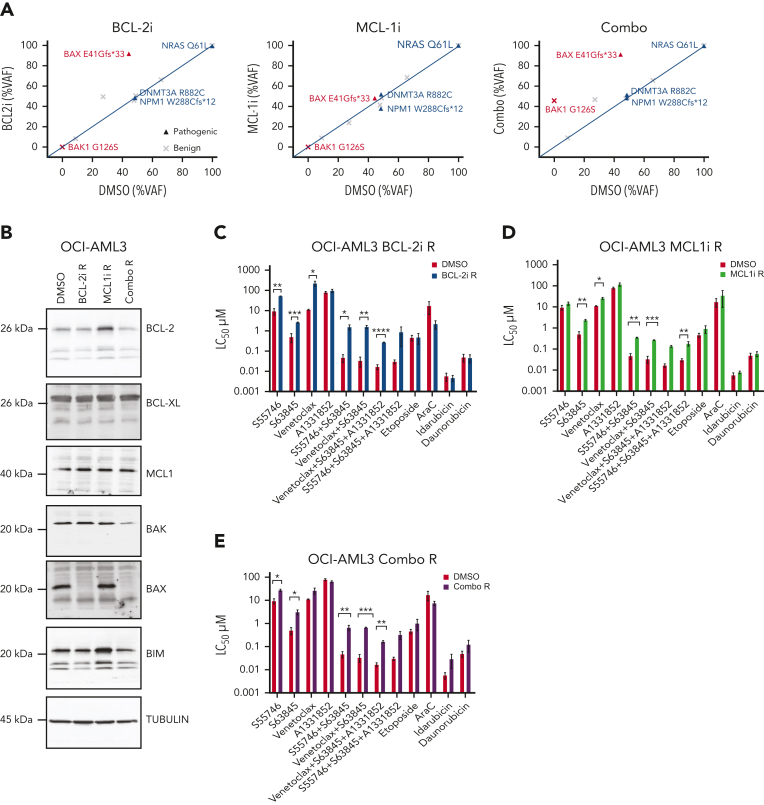
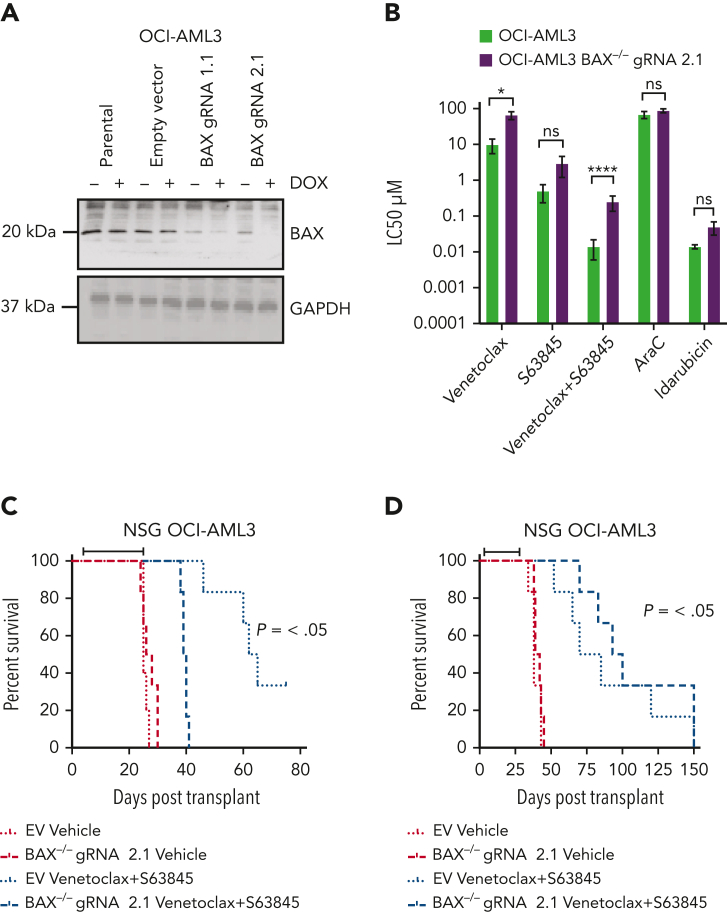
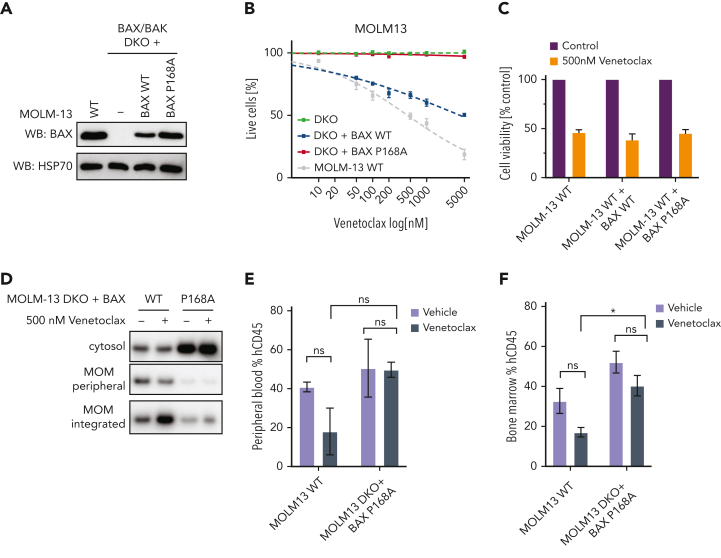
Randomized trials in acute myeloid leukemia (AML) have demonstrated improved survival by the BCL-2 inhibitor venetoclax combined with azacitidine in older patients, and clinical trials are actively exploring the role of venetoclax in combination with intensive chemotherapy in fitter patients with AML. As most patients still develop recurrent disease, improved understanding of relapse mechanisms is needed. We find that 17% of patients relapsing after venetoclax-based therapy for AML have acquired inactivating missense or frameshift/nonsense mutations in the apoptosis effector gene BAX. In contrast, such variants were rare after genotoxic chemotherapy. BAX variants arose within either leukemic or preleukemic compartments, with multiple mutations observed in some patients. In vitro, AML cells with mutated BAX were competitively selected during prolonged exposure to BCL-2 antagonists. In model systems, AML cells rendered deficient for BAX, but not its close relative BAK, displayed resistance to BCL-2 targeting, whereas sensitivity to conventional chemotherapy was variable. Acquired mutations in BAX during venetoclax-based therapy represent a novel mechanism of resistance to BH3-mimetics and a potential barrier to the long-term efficacy of drugs targeting BCL-2 in AML.
© 2023 by The American Society of Hematology. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: S.B. is an employee of Servier. A.W.R., D.C.S.H., A.H.W., N.S.A., and D.M.M are current employees, whereas M.A.D. is a former employee of the Walter and Eliza Hall Institute of Medical Research, which has received milestone and royalty payments related to venetoclax. A.W.R is an inventor on a patent related to venetoclax assigned to AbbVie and Genentech. D.C.S.H. has received research funding from Genentech. A.W.R., D.C.S.H., and A.H.W. have received research funding from Servier and AbbVie. The remaining authors declare no competing financial interests.
Figures
Comment in
-
Acquired BAX mutations in AML.Blood. 2023 Feb 9;141(6):562-564. doi: 10.1182/blood.2022018508. Blood. 2023. PMID: 36757728 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
