

BDK inhibition acts as a catabolic switch to mimic fasting and improve metabolism in mice
- PMID: 36220546
- PMCID: PMC9589198
- DOI: 10.1016/j.molmet.2022.101611
BDK inhibition acts as a catabolic switch to mimic fasting and improve metabolism in mice
Abstract
Objective: Branched chain amino acid (BCAA) catabolic defects are implicated to be causal determinates of multiple diseases. This work aimed to better understand how enhancing BCAA catabolism affected metabolic homeostasis as well as the mechanisms underlying these improvements.
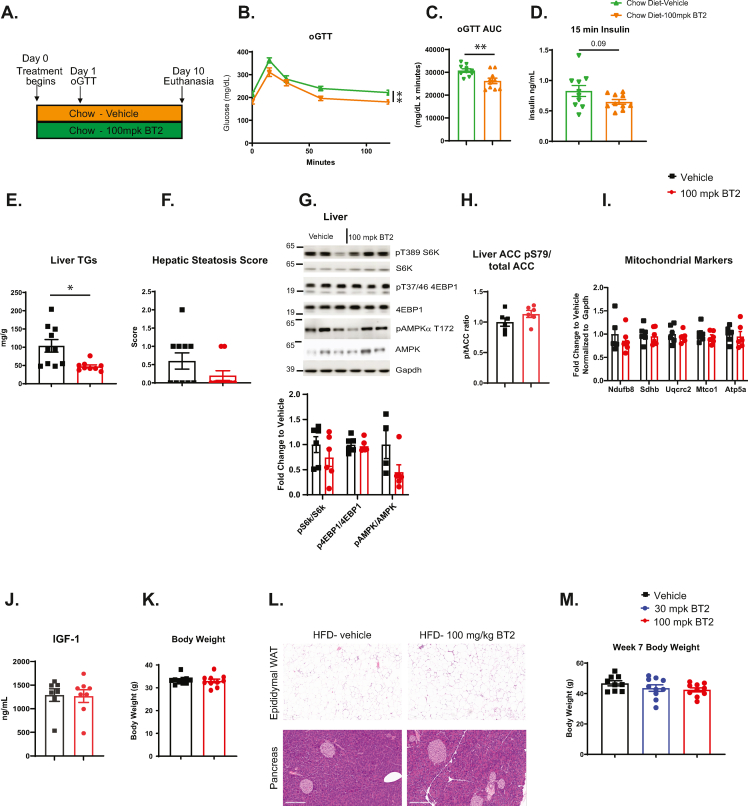
Methods: The rate limiting step of BCAA catabolism is the irreversible decarboxylation by the branched chain ketoacid dehydrogenase (BCKDH) enzyme complex, which is post-translationally controlled through phosphorylation by BCKDH kinase (BDK). This study utilized BT2, a small molecule allosteric inhibitor of BDK, in multiple mouse models of metabolic dysfunction and NAFLD including the high fat diet (HFD) model with acute and chronic treatment paradigms, the choline deficient and methionine minimal high fat diet (CDAHFD) model, and the low-density lipoprotein receptor null mouse model (Ldlr-/-). shRNA was additionally used to knock down BDK in liver to elucidate liver-specific effects of BDK inhibition in HFD-fed mice.
Results: A rapid improvement in insulin sensitivity was observed in HFD-fed and lean mice after BT2 treatment. Resistance to steatosis was assessed in HFD-fed mice, CDAHFD-fed mice, and Ldlr-/- mice. In all cases, BT2 treatment reduced steatosis and/or inflammation. Fasting and refeeding demonstrated a lack of response to feeding-induced changes in plasma metabolites including insulin and beta-hydroxybutyrate and hepatic gene changes in BT2-treated mice. Mechanistically, BT2 treatment acutely altered the expression of genes involved in fatty acid oxidation and lipogenesis in liver, and upstream regulator analysis suggested that BT2 treatment activated PPARα. However, BT2 did not directly activate PPARα in vitro. Conversely, shRNA-AAV-mediated knockdown of BDK specifically in liver in vivo did not demonstrate any effects on glycemia, steatosis, or PPARα-mediated gene expression in mice.
Conclusions: These data suggest that BT2 treatment acutely improves metabolism and liver steatosis in multiple mouse models. While many molecular changes occur in liver in BT2-treated mice, these changes were not observed in mice with AAV-mediated shRNA knockdown of BDK. All together, these data suggest that systemic BDK inhibition is required to improve metabolism and steatosis by prolonging a fasting signature in a paracrine manner. Therefore, BCAA may act as a "fed signal" to promote nutrient storage and reduced systemic BCAA levels as shown in this study via BDK inhibition may act as a "fasting signal" to prolong the catabolic state.
Keywords: BCAA; Diabetes; Metabolic syndrome; Metabolism; NAFLD.
Copyright © 2022 The Author(s). Published by Elsevier GmbH.. All rights reserved.
Figures












References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
