

C9orf72 functions in the nucleus to regulate DNA damage repair
- PMID: 36220889
- PMCID: PMC9984389
- DOI: 10.1038/s41418-022-01074-0
C9orf72 functions in the nucleus to regulate DNA damage repair
Abstract
The hexanucleotide GGGGCC repeat expansion in the intronic region of C9orf72 is the most common cause of Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). The repeat expansion-generated toxic RNAs and dipeptide repeats (DPRs) including poly-GR, have been extensively studied in neurodegeneration. Moreover, haploinsufficiency has been implicated as a disease mechanism but how C9orf72 deficiency contributes to neurodegeneration remains unclear. Here, we show that C9orf72 deficiency exacerbates poly-GR-induced neurodegeneration by attenuating non-homologous end joining (NHEJ) repair. We demonstrate that C9orf72 localizes to the nucleus and is rapidly recruited to sites of DNA damage. C9orf72 deficiency resulted in impaired NHEJ repair through attenuated DNA-PK complex assembly and DNA damage response (DDR) signaling. In mouse models, we found that C9orf72 deficiency exacerbated poly-GR-induced neuronal loss, glial activation, and neuromuscular deficits. Furthermore, DNA damage accumulated in C9orf72-deficient neurons that expressed poly-GR, resulting in excessive activation of PARP-1. PARP-1 inhibition rescued neuronal death in cultured neurons treated with poly-GR peptides. Together, our results support a pathological mechanism where C9orf72 haploinsufficiency synergizes with poly-GR-induced DNA double-strand breaks to exacerbate the accumulation of DNA damage and PARP-1 overactivation in C9orf72 ALS/FTD patients.
© 2022. The Author(s), under exclusive licence to ADMC Associazione Differenziamento e Morte Cellulare.
Conflict of interest statement
The authors declare no competing interests.
Figures
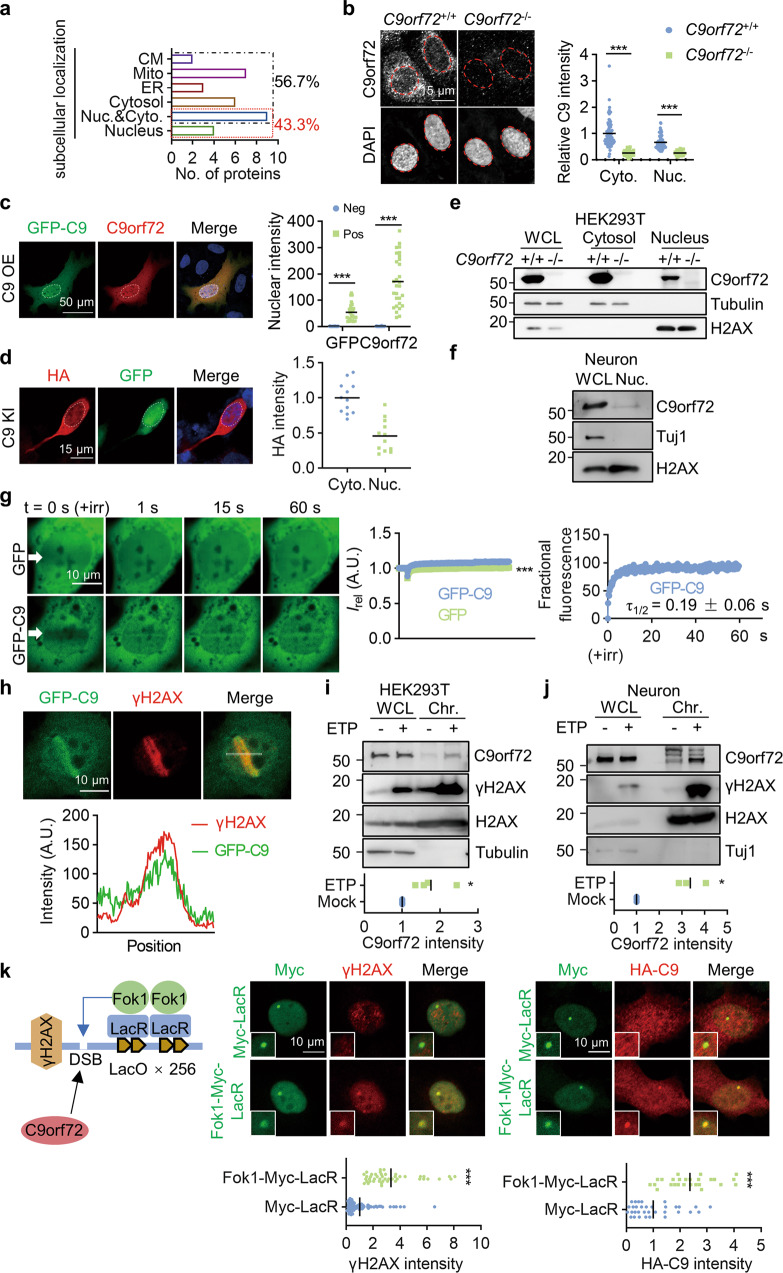
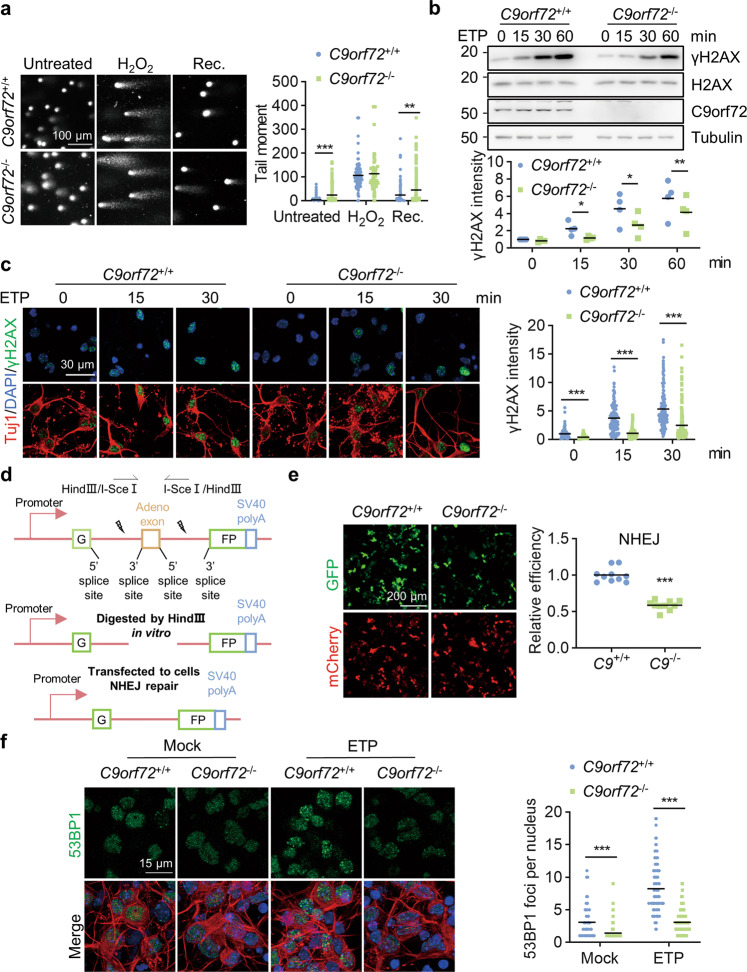
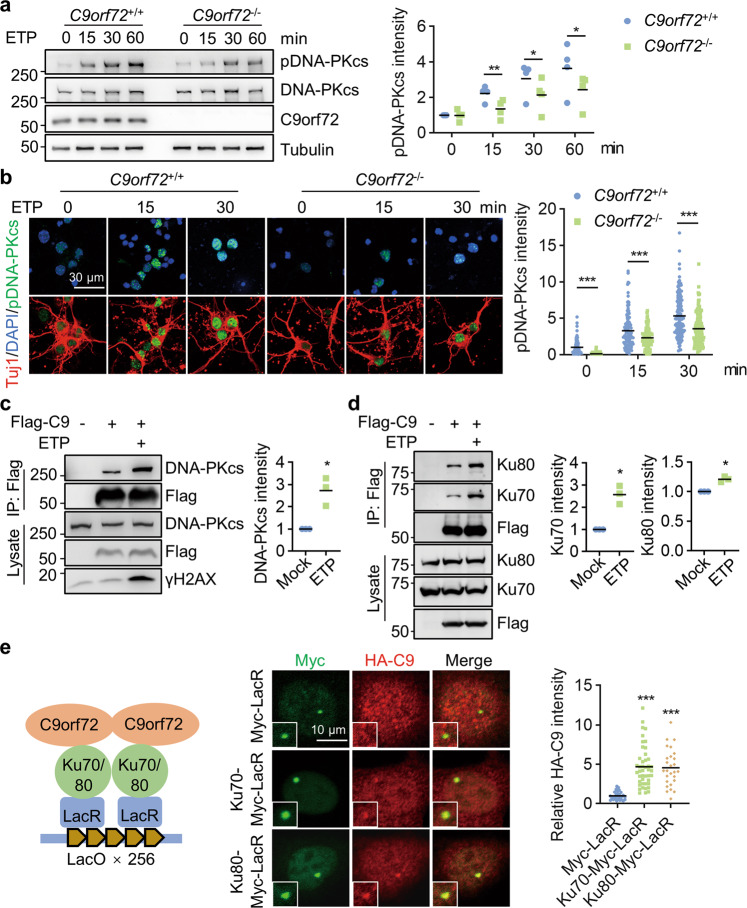
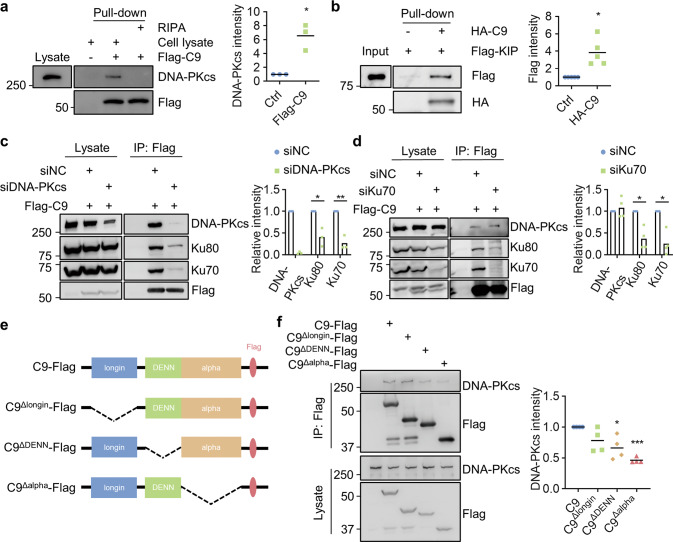
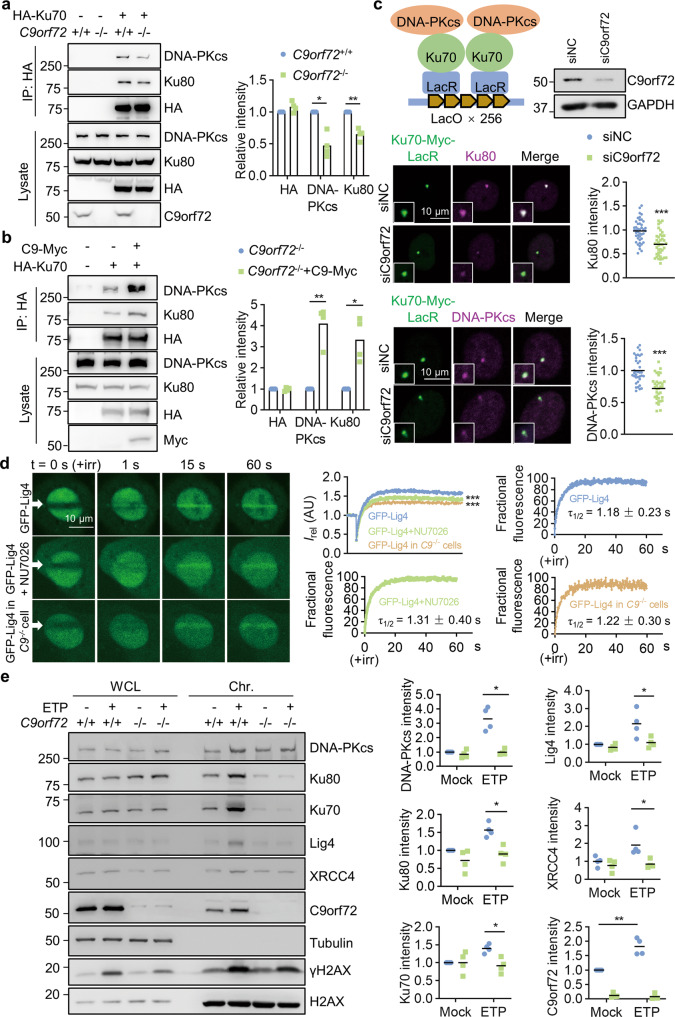
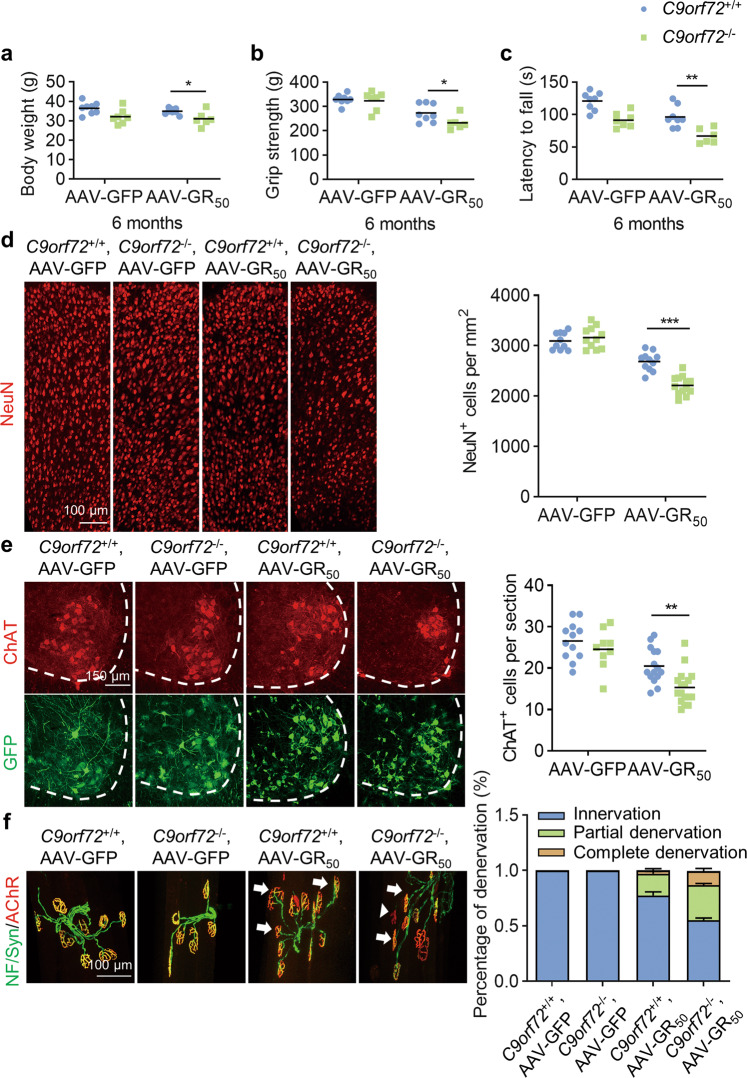
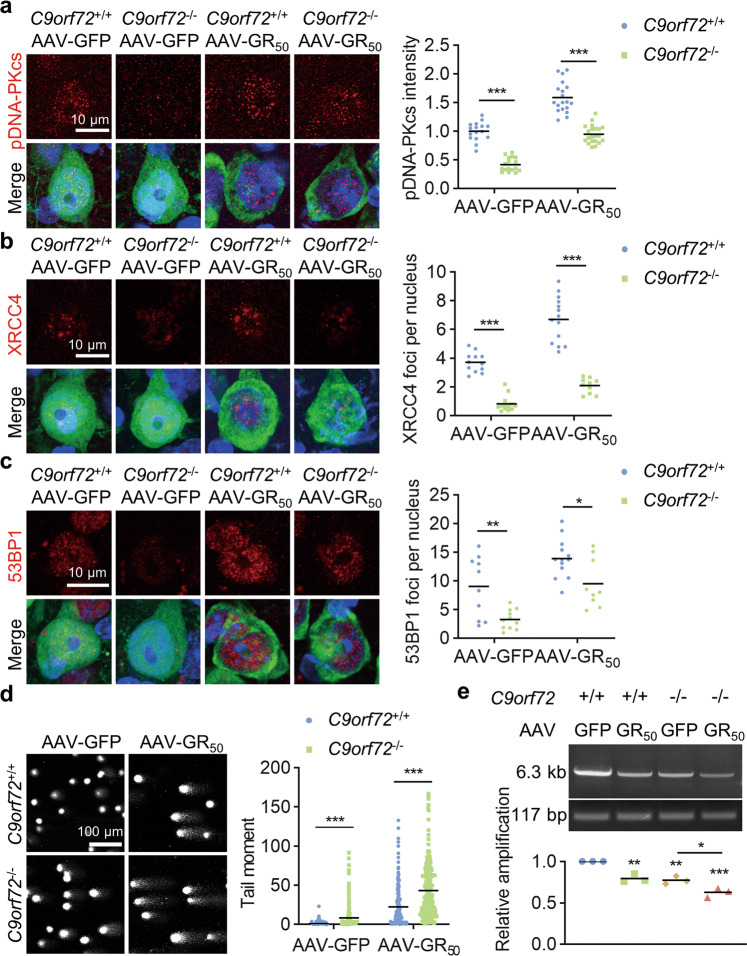
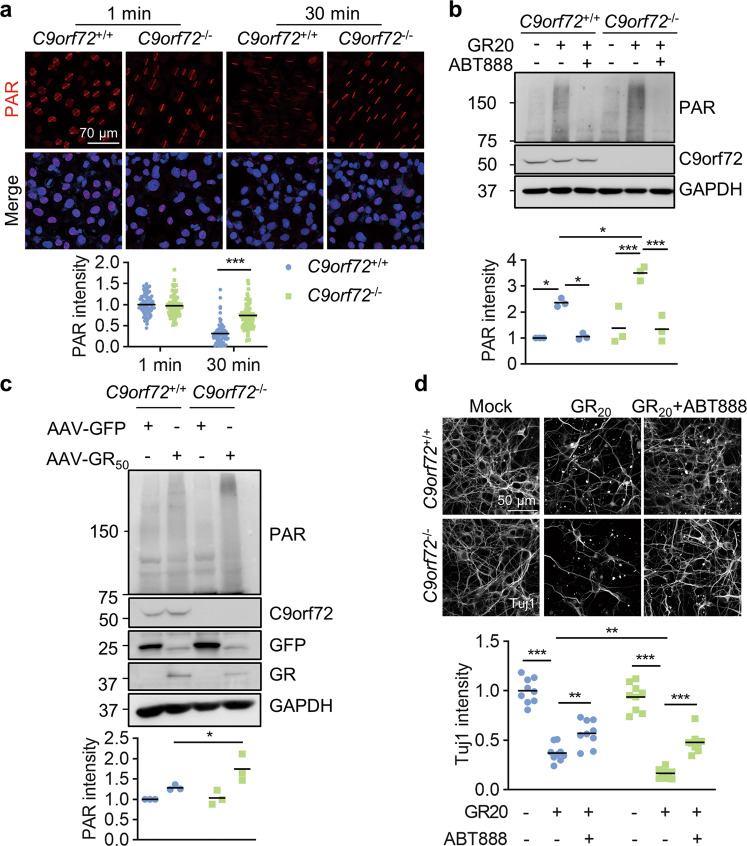
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
