

TP53 R249S mutation in hepatic organoids captures the predisposing cancer risk
- PMID: 36221953
- PMCID: PMC10086078
- DOI: 10.1002/hep.32802
TP53 R249S mutation in hepatic organoids captures the predisposing cancer risk
Abstract
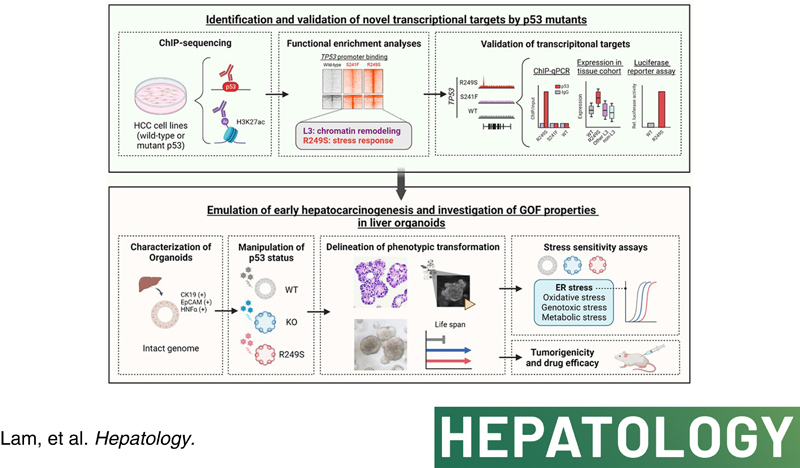
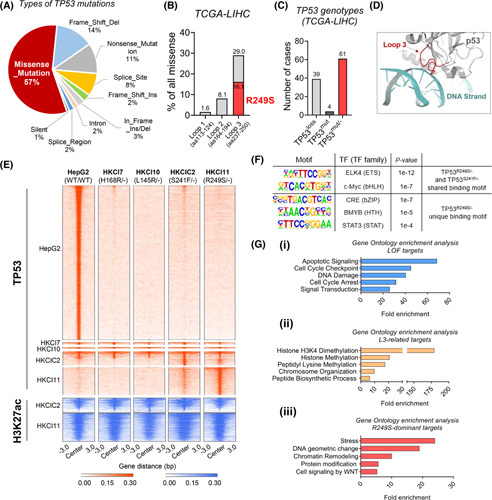
Background and aims: Major genomic drivers of hepatocellular carcinoma (HCC) are nowadays well recognized, although models to establish their roles in human HCC initiation remain scarce. Here, we used human liver organoids in experimental systems to mimic the early stages of human liver carcinogenesis from the genetic lesions of TP53 loss and L3 loop R249S mutation. In addition, chromatin immunoprecipitation sequencing (ChIP-seq) of HCC cell lines shed important functional insights into the initiation of HCC consequential to the loss of tumor-suppressive function from TP53 deficiency and gain-of-function activities from mutant p53.
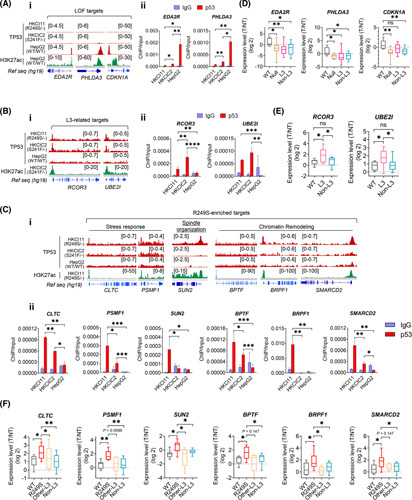
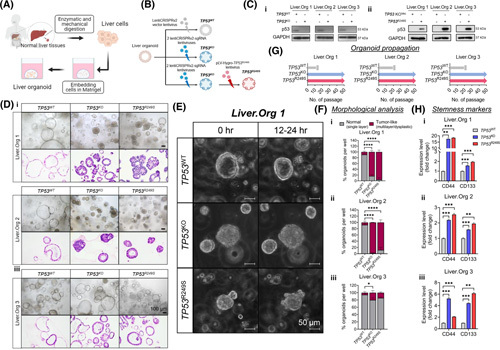
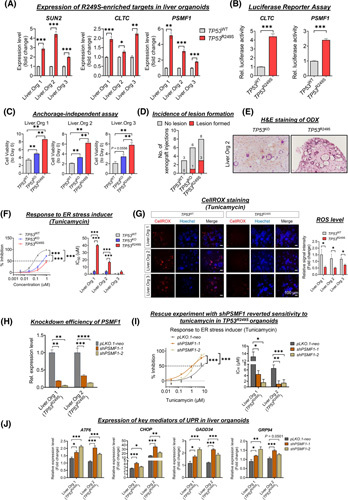
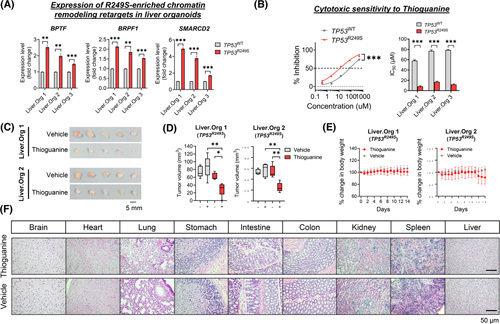
Approach and results: Human liver organoids were generated from surgical nontumor liver tissues. CRISPR knockout of TP53 in liver organoids consistently demonstrated tumor-like morphological changes, increased in stemness and unrestricted in vitro propagation. To recapitulate TP53 status in human HCC, we overexpressed mutant R249S in TP53 knockout organoids. A spontaneous increase in tumorigenic potentials and bona fide HCC histology in xenotransplantations were observed. ChIP-seq analysis of HCC cell lines underscored gain-of-function properties from L3 loop p53 mutants in chromatin remodeling and overcoming extrinsic stress. More importantly, direct transcriptional activation of PSMF1 by mutant R249S could increase organoid resistance to endoplasmic reticulum stress, which was readily abrogated by PSMF1 knockdown in rescue experiments. In a patient cohort of primary HCC tumors and genome-edited liver organoids, quantitative polymerase chain reaction corroborated ChIP-seq findings and verified preferential genes modulated by L3 mutants, especially those enriched by R249S.
Conclusions: We showed differential tumorigenic effects from TP53 loss and L3 mutations, which together confer normal hepatocytes with early clonal advantages and prosurvival functions.
Copyright © 2023 The Author(s). Published by Wolters Kluwer Health, Inc.
Conflict of interest statement
This work was supported by the Hong Kong Research Grants Council Area of Excellence Scheme (Ref. AoE/M‐401/20), Research Impact Fund (Ref. R4017‐18) and internal funds from CUHK. This research is also supported in part by a National Cancer Institute fund of the National Institutes of Health (No. R01CA229836).
Nothing to report.
Figures
References
-
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
