

The Role of Molossidae and Vespertilionidae in Shaping the Diversity of Alphacoronaviruses in the Americas
- PMID: 36222689
- PMCID: PMC9769993
- DOI: 10.1128/spectrum.03143-22
The Role of Molossidae and Vespertilionidae in Shaping the Diversity of Alphacoronaviruses in the Americas
Abstract
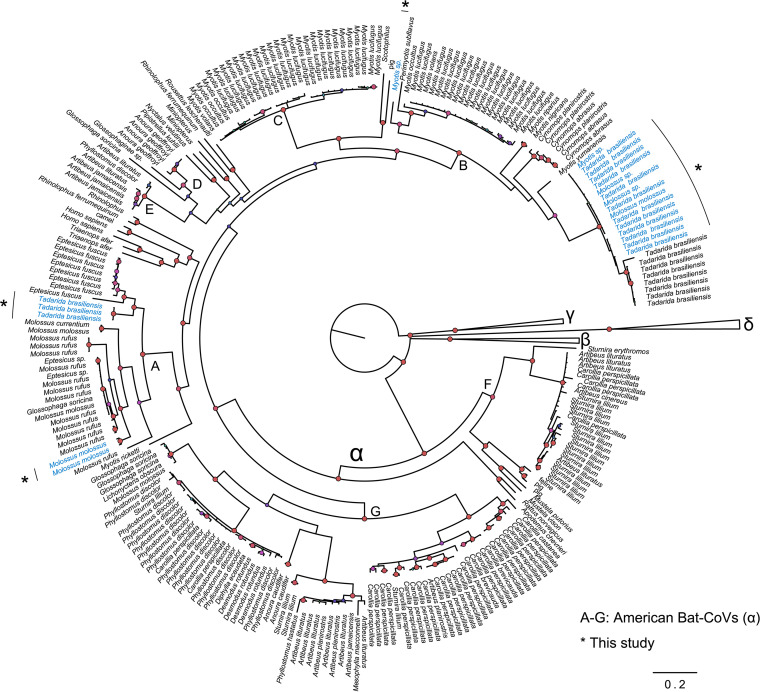
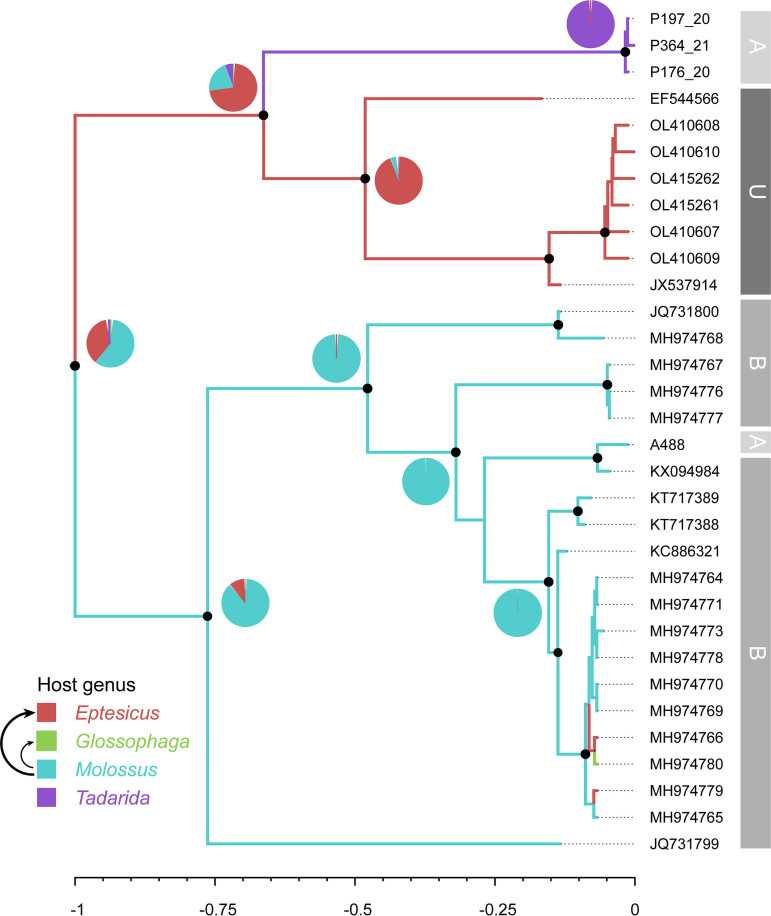
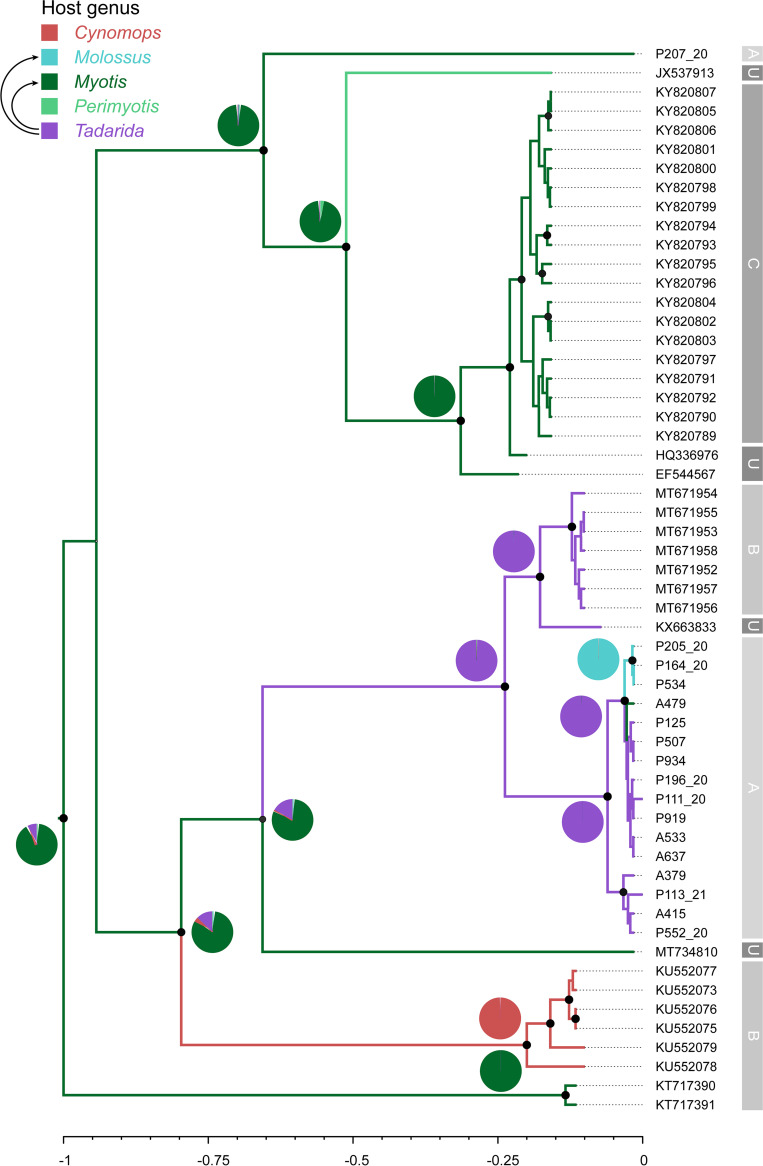
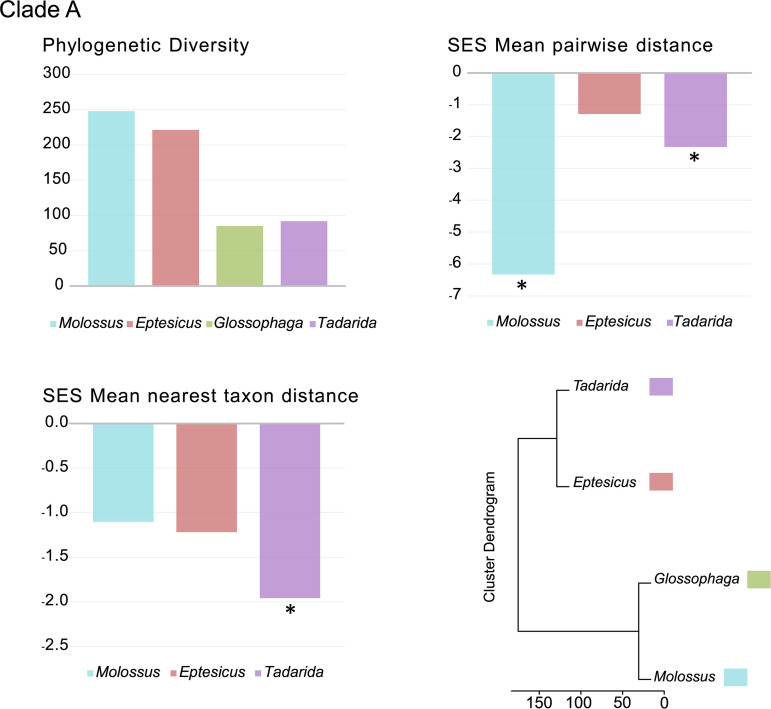
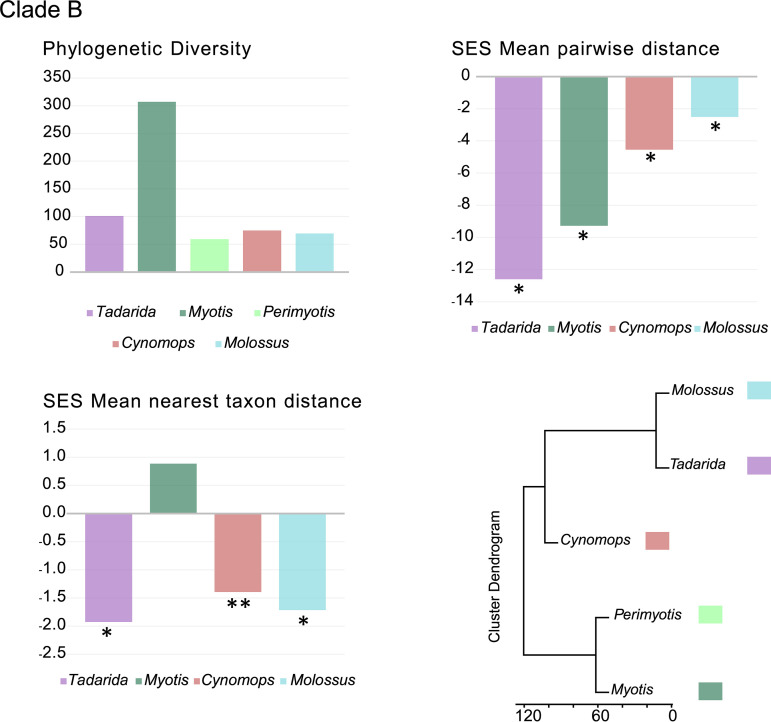
Bats are reservoirs of diverse coronaviruses (CoVs), including progenitors of severe acute respiratory syndrome CoV (SARS-CoV) and SARS-CoV-2. In the Americas, there is a contrast between alphacoronaviruses (alphaCoVs) and betaCoVs: while cospeciation prevails in the latter, alphaCoV evolution is dominated by deep and recent host switches. AlphaCoV lineages are maintained by two different bat family groups, Phyllostomidae and Vespertilionidae plus Molossidae. In this study, we used a Bayesian framework to analyze the process of diversification of the lineages maintained by Molossidae and Vespertilionidae, adding novel CoV sequences from Argentina. We provide evidence that the observed CoV diversity in these two bat families is shaped by their geographic distribution and that CoVs exhibit clustering at the level of bat genera. We discuss the causes of the cocirculation of two independent clades in Molossus and Tadarida as well as the role of Myotis as the ancestral host and a major evolutionary reservoir of alphaCoVs across the continent. Although more CoV sampling efforts are needed, these findings contribute to a better knowledge of the diversity of alphaCoVs and the links between bat host species. IMPORTANCE Bats harbor the largest diversity of coronaviruses among mammals. In the Americas, seven alphacoronavirus lineages circulate among bats. Three of these lineages are shared by members of two bat families: Vespertilionidae and Molossidae. Uncovering the relationships between these coronaviruses can help us to understand patterns of cross-species transmission and, ultimately, which hosts are more likely to be involved in spillover events. We found that two different lineages cocirculate among the bat genera Molossus and Tadarida, which share roosts and have common viral variants. The bat genus Myotis functions as a reservoir of coronavirus diversity and, as such, is a key host. Although there were some spillovers recorded, there is a strong host association, showing that once a successful host jump takes place, it is transmitted onward to members of the same bat genus.
Keywords: Molossidae; Vespertilionidae; bats; coronavirus; cross-species transmission; host shift; phylogeny; spillover; virus.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
