

Genetic and environmental drivers of large-scale epigenetic variation in Thlaspi arvense
- PMID: 36223399
- PMCID: PMC9591053
- DOI: 10.1371/journal.pgen.1010452
Genetic and environmental drivers of large-scale epigenetic variation in Thlaspi arvense
Abstract
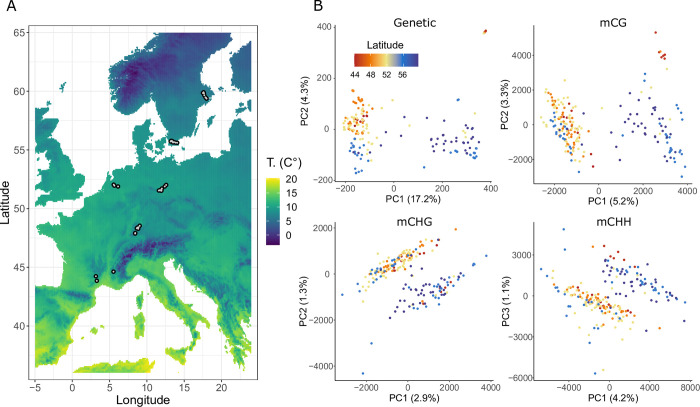
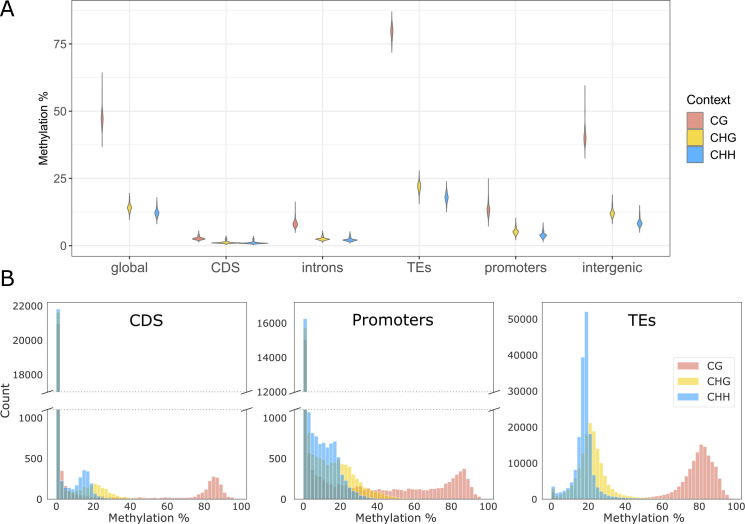
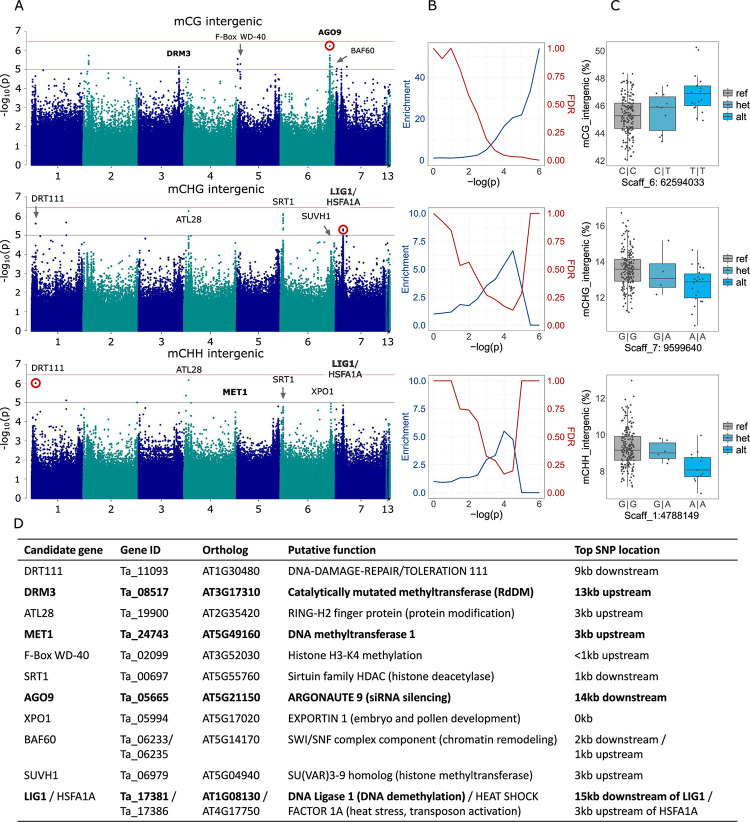
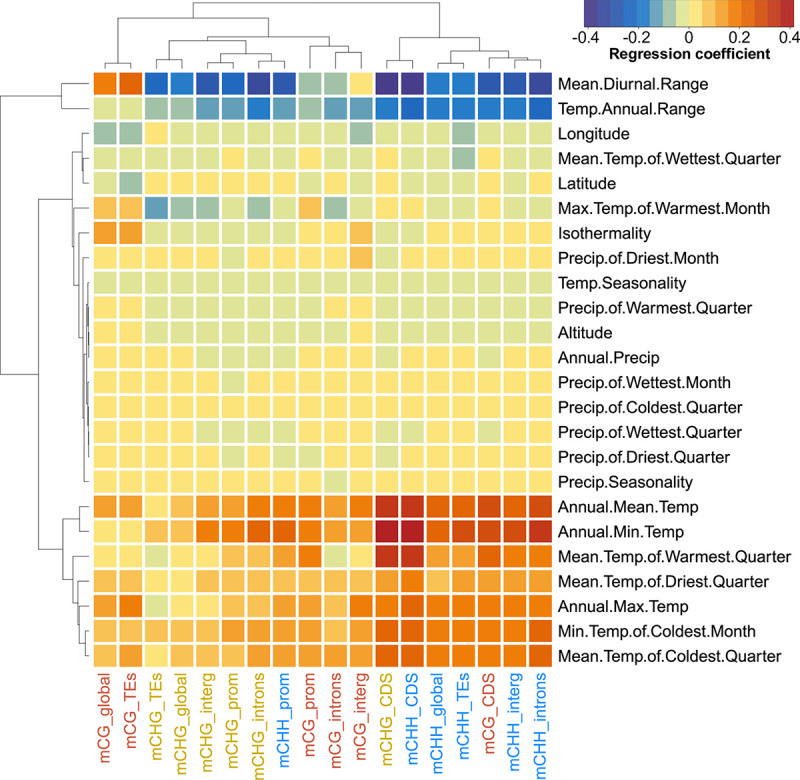
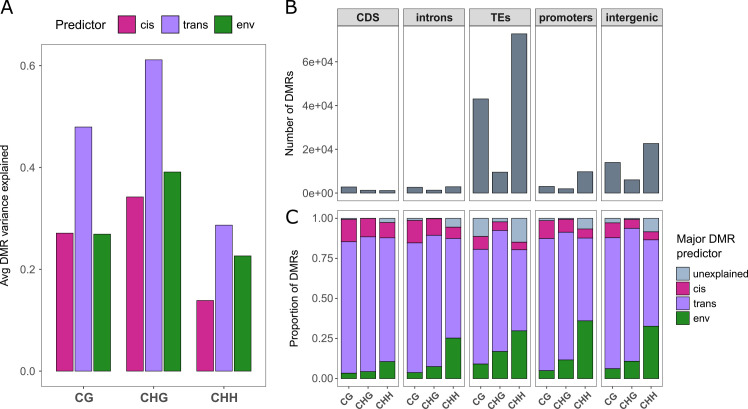
Natural plant populations often harbour substantial heritable variation in DNA methylation. However, a thorough understanding of the genetic and environmental drivers of this epigenetic variation requires large-scale and high-resolution data, which currently exist only for a few model species. Here, we studied 207 lines of the annual weed Thlaspi arvense (field pennycress), collected across a large latitudinal gradient in Europe and propagated in a common environment. By screening for variation in DNA sequence and DNA methylation using whole-genome (bisulfite) sequencing, we found significant epigenetic population structure across Europe. Average levels of DNA methylation were strongly context-dependent, with highest DNA methylation in CG context, particularly in transposable elements and in intergenic regions. Residual DNA methylation variation within all contexts was associated with genetic variants, which often co-localized with annotated methylation machinery genes but also with new candidates. Variation in DNA methylation was also significantly associated with climate of origin, with methylation levels being lower in colder regions and in more variable climates. Finally, we used variance decomposition to assess genetic versus environmental associations with differentially methylated regions (DMRs). We found that while genetic variation was generally the strongest predictor of DMRs, the strength of environmental associations increased from CG to CHG and CHH, with climate-of-origin as the strongest predictor in about one third of the CHH DMRs. In summary, our data show that natural epigenetic variation in Thlaspi arvense is significantly associated with both DNA sequence and environment of origin, and that the relative importance of the two factors strongly depends on the sequence context of DNA methylation. T. arvense is an emerging biofuel and winter cover crop; our results may hence be relevant for breeding efforts and agricultural practices in the context of rapidly changing environmental conditions.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Becker C, Hagmann J, Müller J, Koenig D, Stegle O, Borgwardt K, et al. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature. 2011. Dec;480(7376):245–9. - PubMed
-
- He Y, Li Z. Epigenetic Environmental Memories in Plants: Establishment, Maintenance, and Reprogramming. Trends in Genetics. 2018. Aug 22; Available from: http://www.sciencedirect.com/science/article/pii/S0168952518301276 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
