

Whole-genome analysis identifies novel drivers and high-risk double-hit events in relapsed/refractory myeloma
- PMID: 36223594
- PMCID: PMC10163277
- DOI: 10.1182/blood.2022017010
Whole-genome analysis identifies novel drivers and high-risk double-hit events in relapsed/refractory myeloma
Abstract
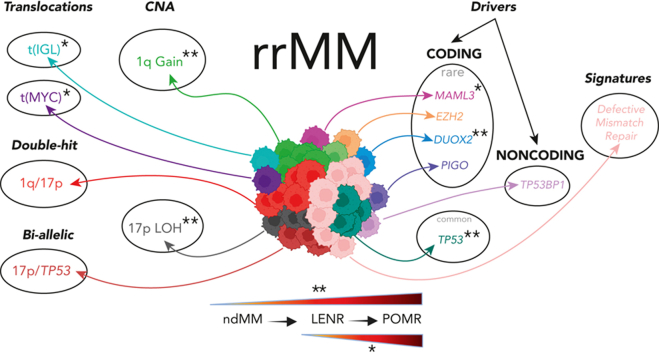
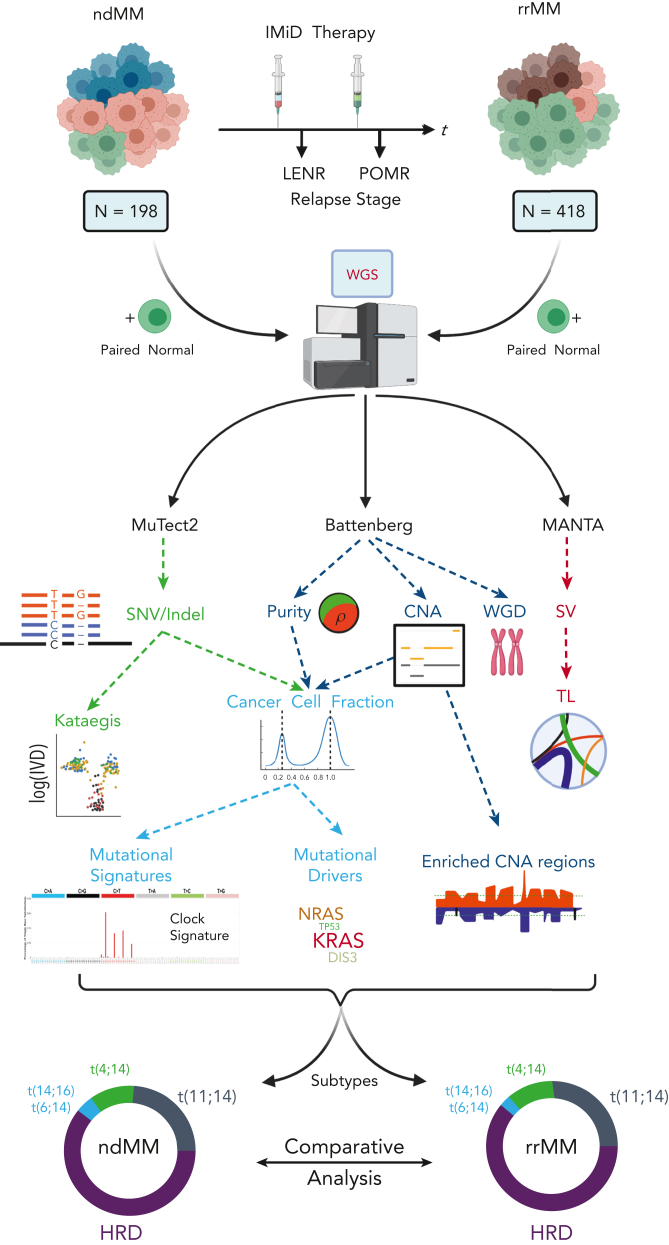
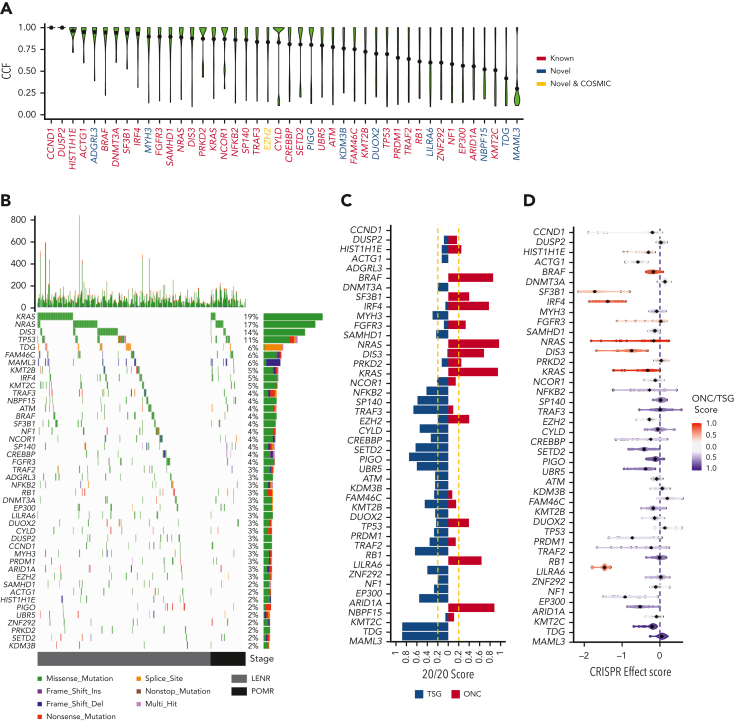
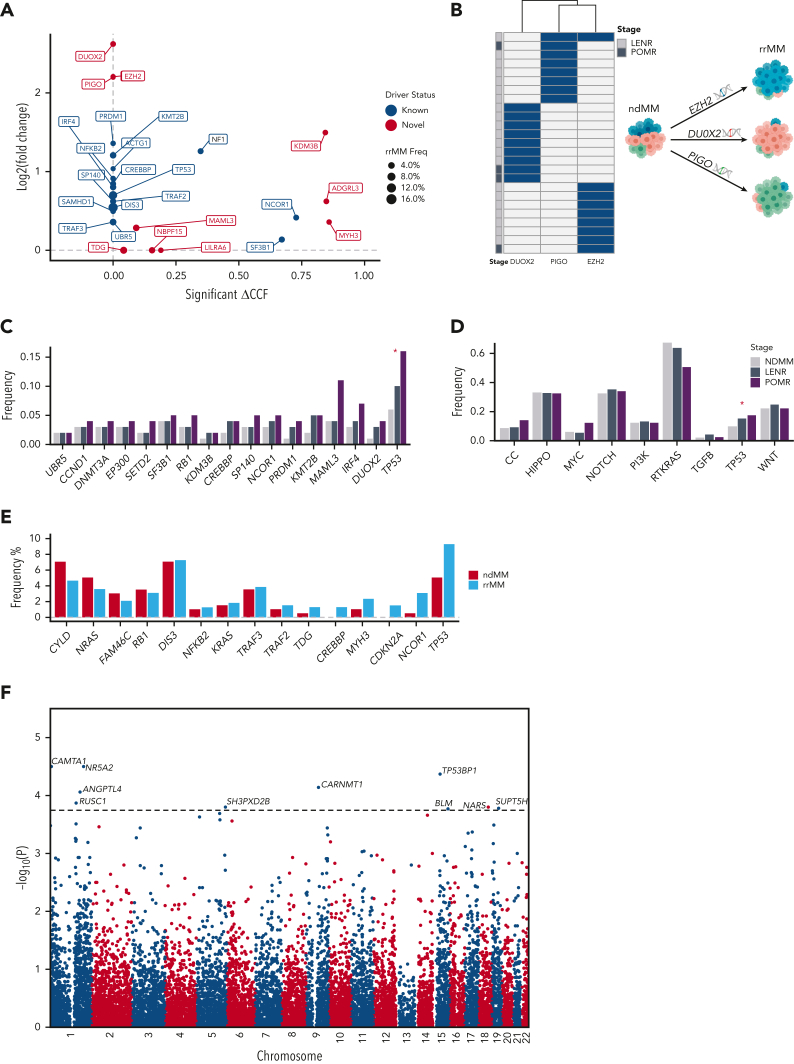
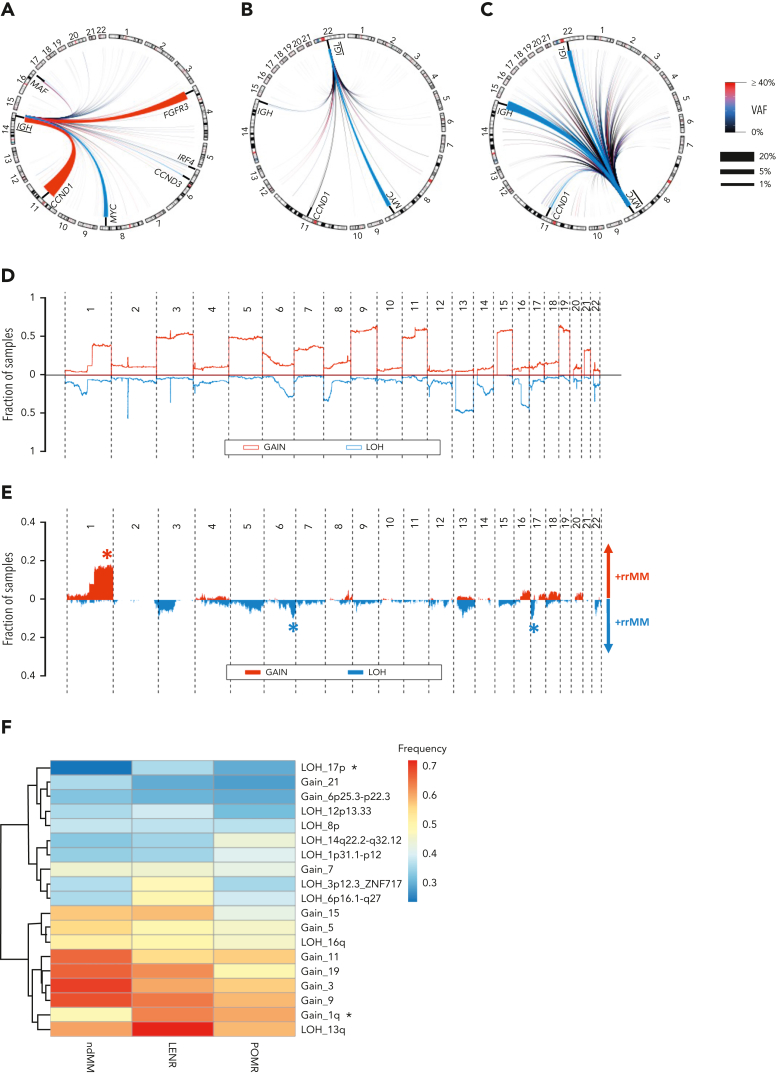
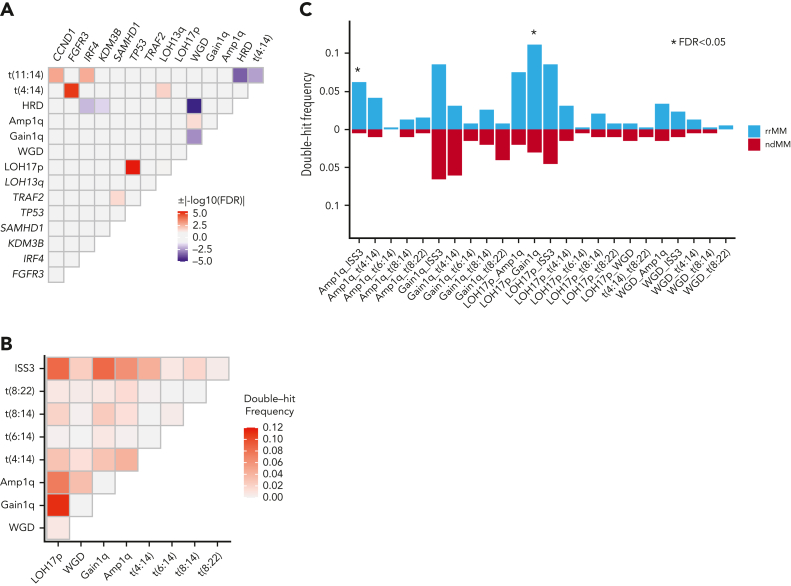
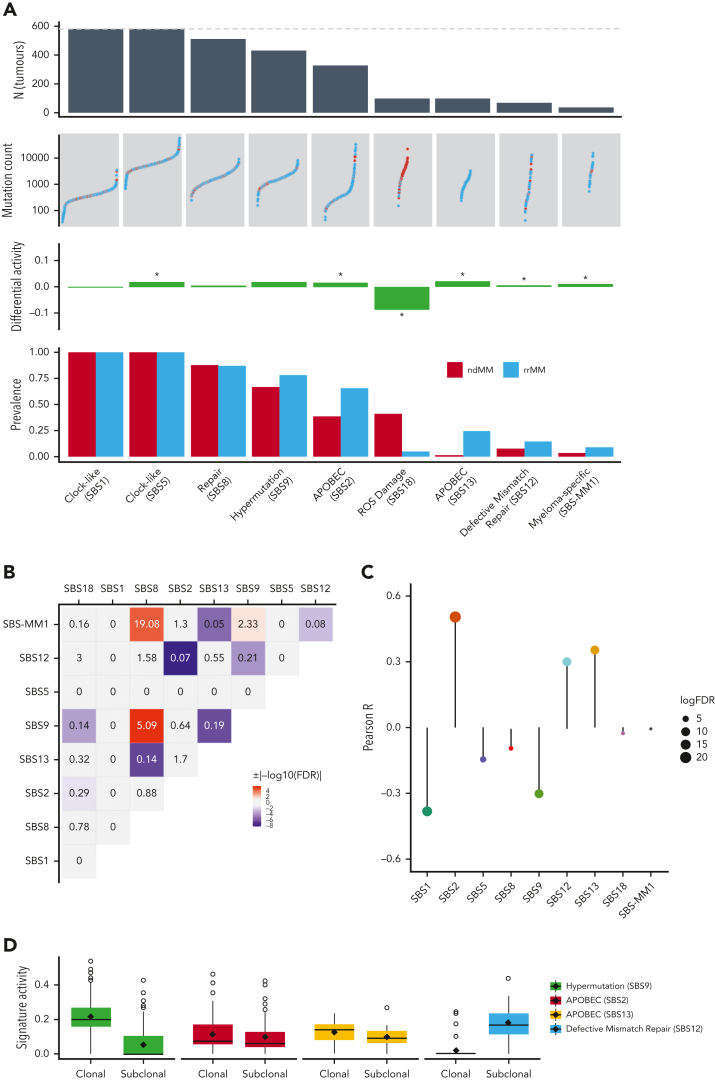
Large-scale analyses of genomic data from patients with newly diagnosed multiple myeloma (ndMM) have been undertaken, however, large-scale analysis of relapsed/refractory MM (rrMM) has not been performed. We hypothesize that somatic variants chronicle the therapeutic exposures and clonal structure of myeloma from ndMM to rrMM stages. We generated whole-genome sequencing (WGS) data from 418 tumors (386 patients) derived from 6 rrMM clinical trials and compared them with WGS from 198 unrelated patients with ndMM in a population-based case-control fashion. We identified significantly enriched events at the rrMM stage, including drivers (DUOX2, EZH2, TP53), biallelic inactivation (TP53), noncoding mutations in bona fide drivers (TP53BP1, BLM), copy number aberrations (CNAs; 1qGain, 17pLOH), and double-hit events (Amp1q-ISS3, 1qGain-17p loss-of-heterozygosity). Mutational signature analysis identified a subclonal defective mismatch repair signature enriched in rrMM and highly active in high mutation burden tumors, a likely feature of therapy-associated expanding subclones. Further analysis focused on the association of genomic aberrations enriched at different stages of resistance to immunomodulatory agent (IMiD)-based therapy. This analysis revealed that TP53, DUOX2, 1qGain, and 17p loss-of-heterozygosity increased in prevalence from ndMM to lenalidomide resistant (LENR) to pomalidomide resistant (POMR) stages, whereas enrichment of MAML3 along with immunoglobulin lambda (IGL) and MYC translocations distinguished POM from the LEN subgroup. Genomic drivers associated with rrMM are those that confer clonal selective advantage under therapeutic pressure. Their role in therapy evasion should be further evaluated in longitudinal patient samples, to confirm these associations with the evolution of clinical resistance and to identify molecular subsets of rrMM for the development of targeted therapies.
© 2023 by The American Society of Hematology. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflicts-of-interest disclosure: Bristol Myers Squibb (BMS) Corporation has employment and equity ownership; funding for data generation, processing, and storage was provided by BMS Corporation; F.T. and A.T. were BMS employees at the time this analysis was conducted; B.W. receives research funding from BMS; and N.A.-P. is a BMS consultant. The remaining authors declare no competing financial interests.
Figures
Comment in
-
The genome of IMiD-refractory myeloma.Blood. 2023 Feb 9;141(6):560-561. doi: 10.1182/blood.2022018461. Blood. 2023. PMID: 36757732 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
