

Spatial maps of T cell receptors and transcriptomes reveal distinct immune niches and interactions in the adaptive immune response
- PMID: 36223726
- PMCID: PMC9745674
- DOI: 10.1016/j.immuni.2022.09.002
Spatial maps of T cell receptors and transcriptomes reveal distinct immune niches and interactions in the adaptive immune response
Abstract
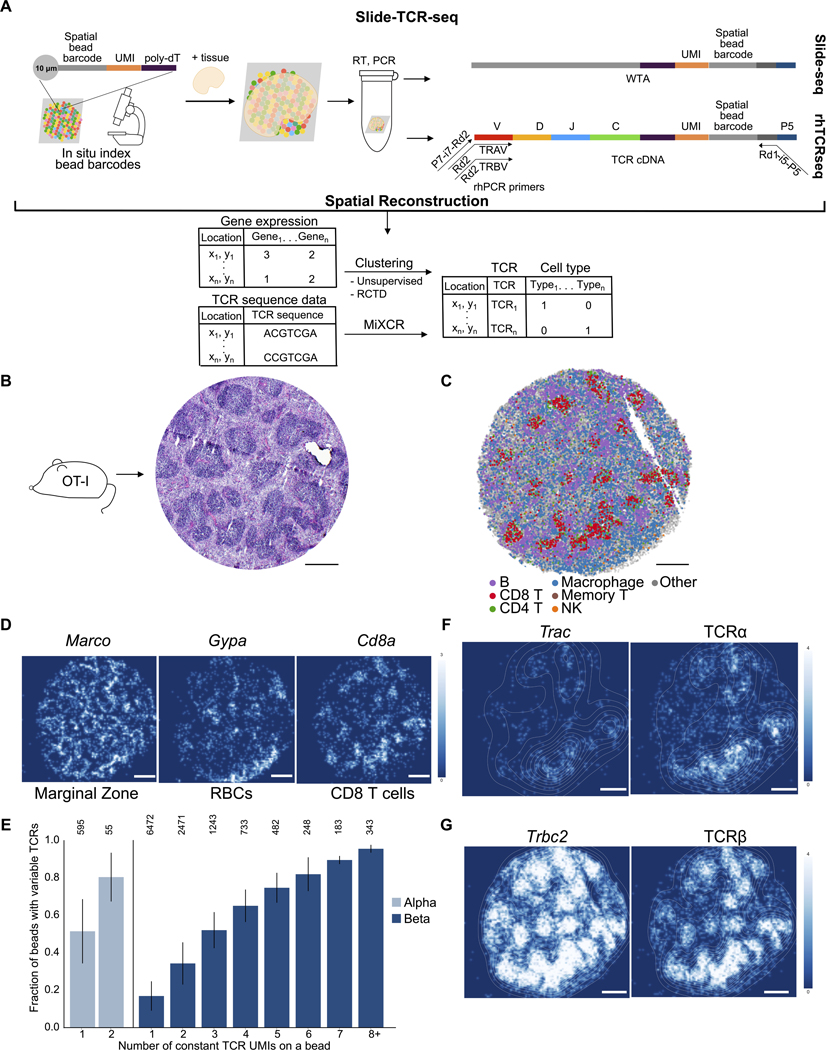
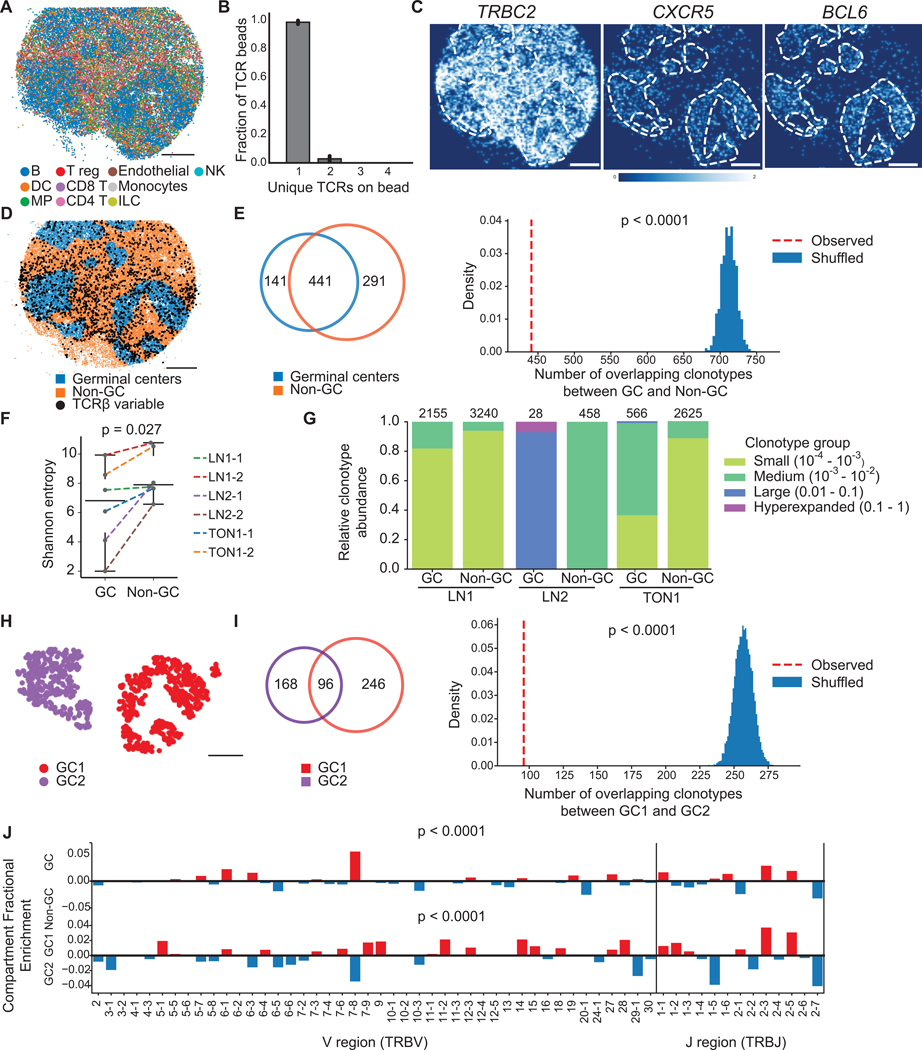
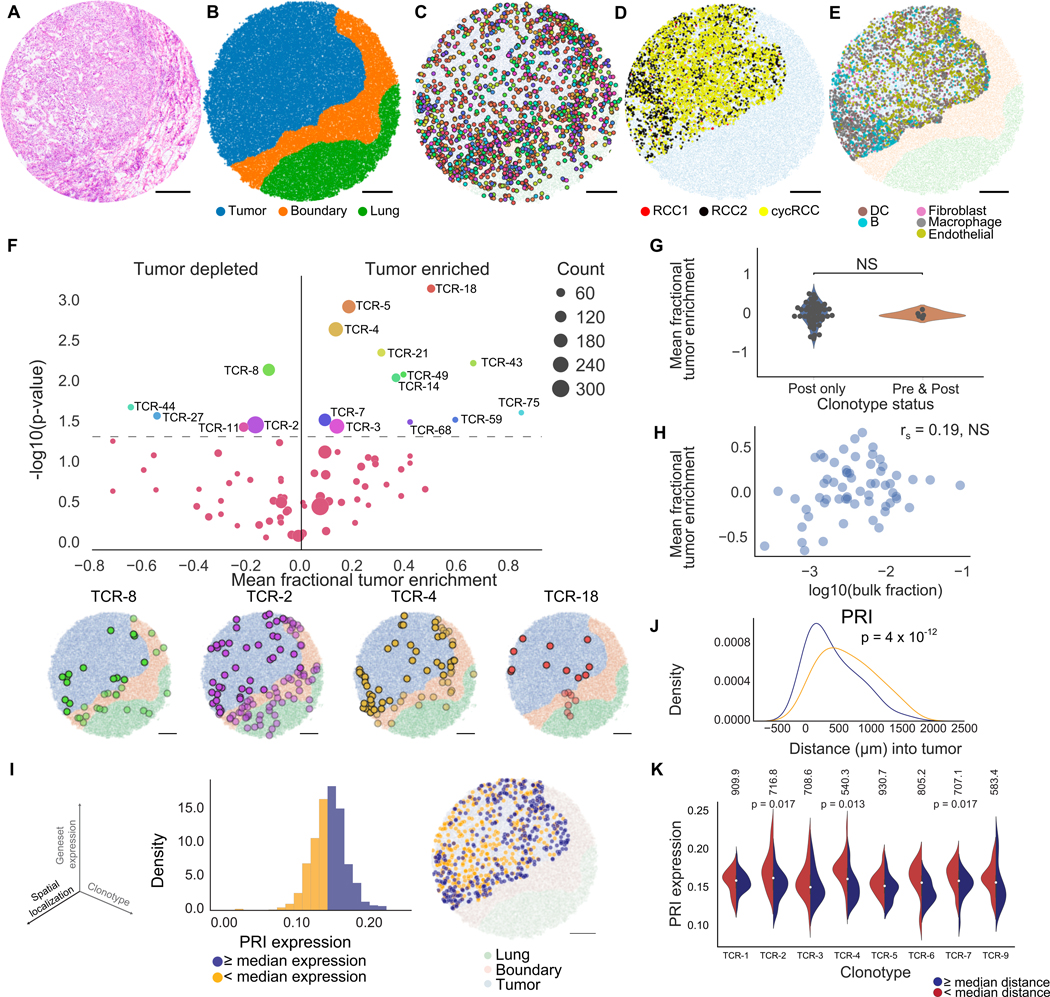
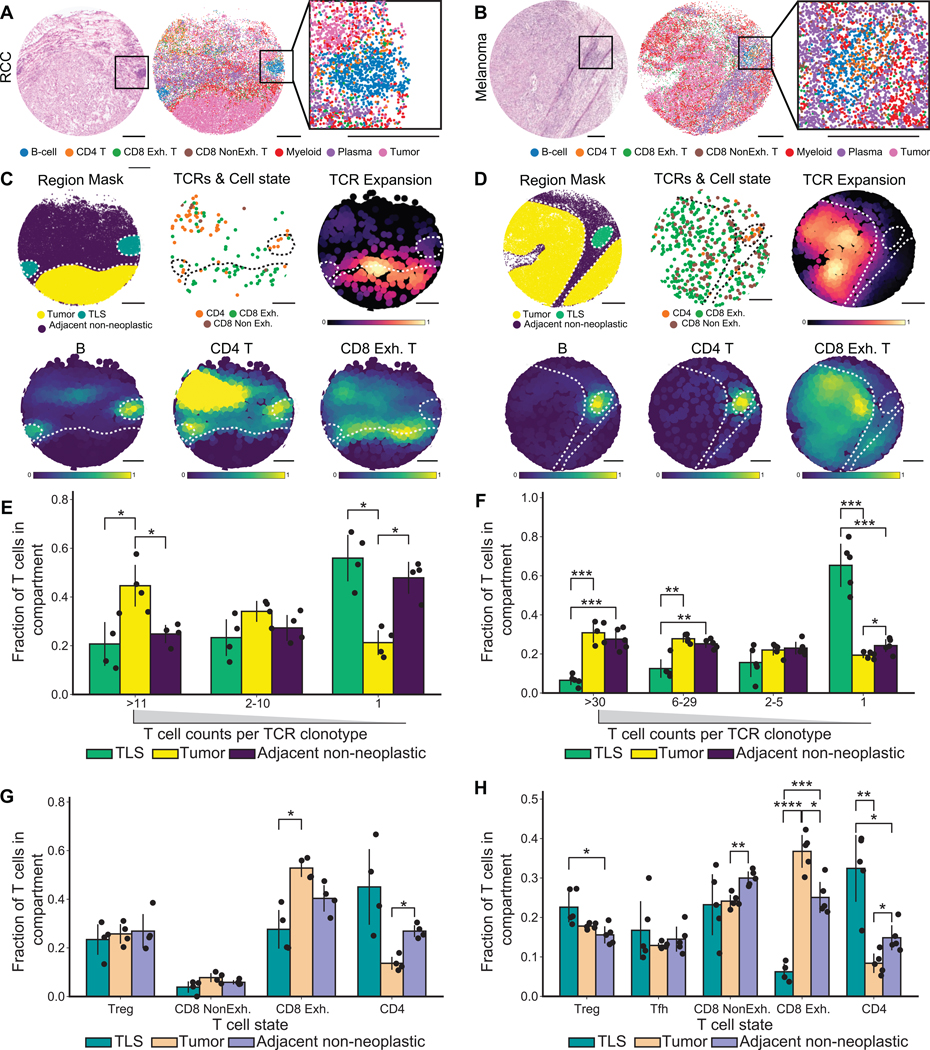
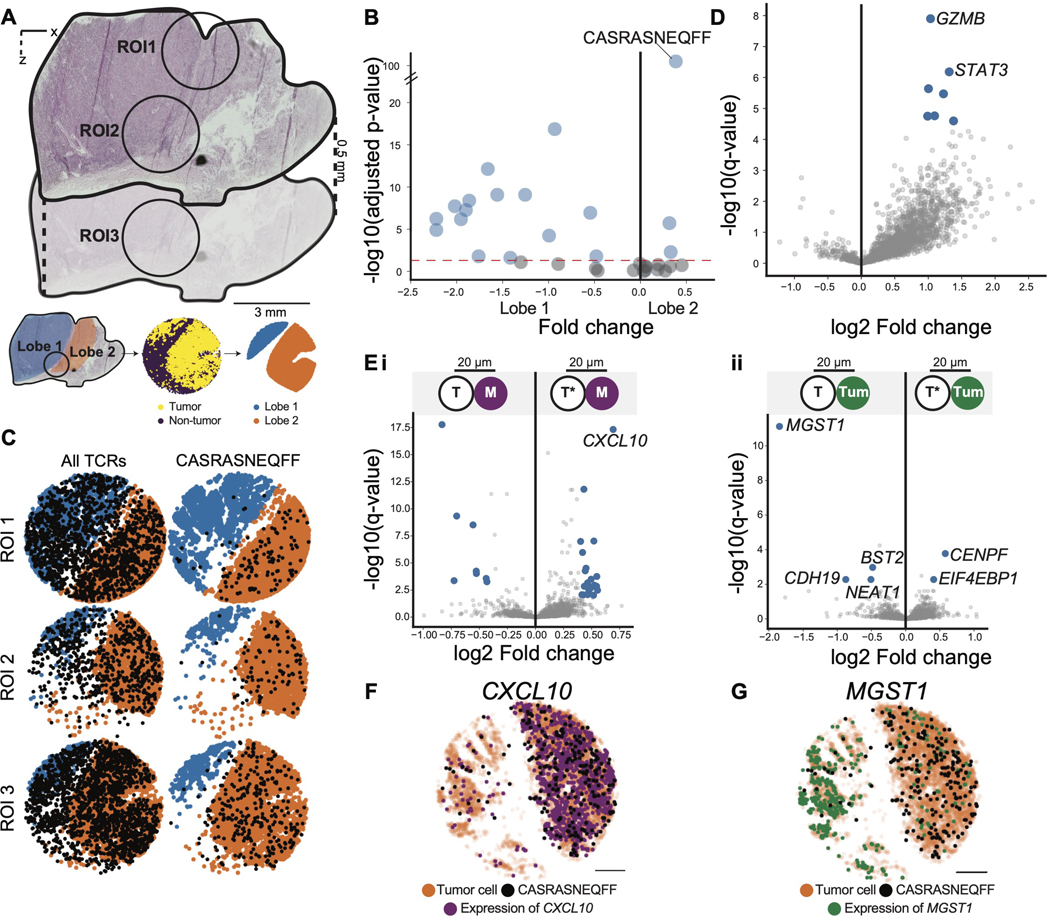
T cells mediate antigen-specific immune responses to disease through the specificity and diversity of their clonotypic T cell receptors (TCRs). Determining the spatial distributions of T cell clonotypes in tissues is essential to understanding T cell behavior, but spatial sequencing methods remain unable to profile the TCR repertoire. Here, we developed Slide-TCR-seq, a 10-μm-resolution method, to sequence whole transcriptomes and TCRs within intact tissues. We confirmed the ability of Slide-TCR-seq to map the characteristic locations of T cells and their receptors in mouse spleen. In human lymphoid germinal centers, we identified spatially distinct TCR repertoires. Profiling T cells in renal cell carcinoma and melanoma specimens revealed heterogeneous immune responses: T cell states and infiltration differed intra- and inter-clonally, and adjacent tumor and immune cells exhibited distinct gene expression. Altogether, our method yields insights into the spatial relationships between clonality, neighboring cell types, and gene expression that drive T cell responses.
Keywords: T cell; T cell receptor; cell interactions; clonotype; immune niches; sequencing; spatial; tertiary lymphoid structures; transcriptomics; tumor infiltrating lymphocytes.
Copyright © 2022 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests F.C., C.J.W., S.L., J.B.I., K.J.L., and S. Li have filed a patent on this work. D.A.B. reports non-financial support from Bristol Myers Squibb; honoraria from LM Education/Exchange Services; advisory board fees from Exelixis and AVEO; personal fees from Charles River Associates, Schlesinger Associates, Imprint Science, Insight Strategy, Trinity Group, Cancer Expert Now, Adnovate Strategies, MDedge, CancerNetwork, Catenion, OncLive, Cello Health BioConsulting, PWW Consulting, Haymarket Medical Network, Aptitude Health, and AbbVie; and research support from Exelixis, outside of the submitted work. C.J.W. holds equity in BioNTech, Inc. K.J.L. holds equity in Standard BioTools Inc. (formerly Fluidigm Corporation). F.C. is a paid consultant for Atlas Bio.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
