

Predicting the structure of large protein complexes using AlphaFold and Monte Carlo tree search
- PMID: 36224222
- PMCID: PMC9556563
- DOI: 10.1038/s41467-022-33729-4
Predicting the structure of large protein complexes using AlphaFold and Monte Carlo tree search
Abstract
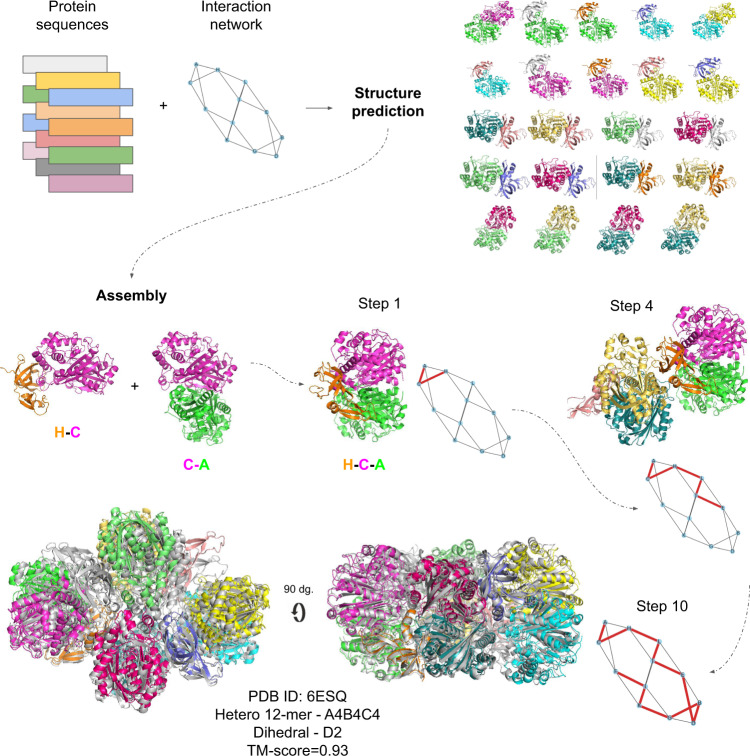
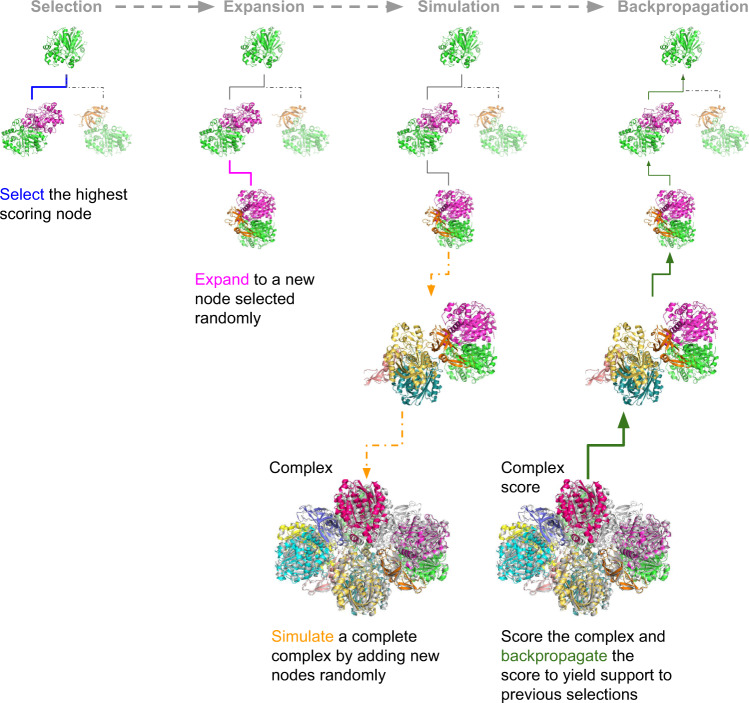
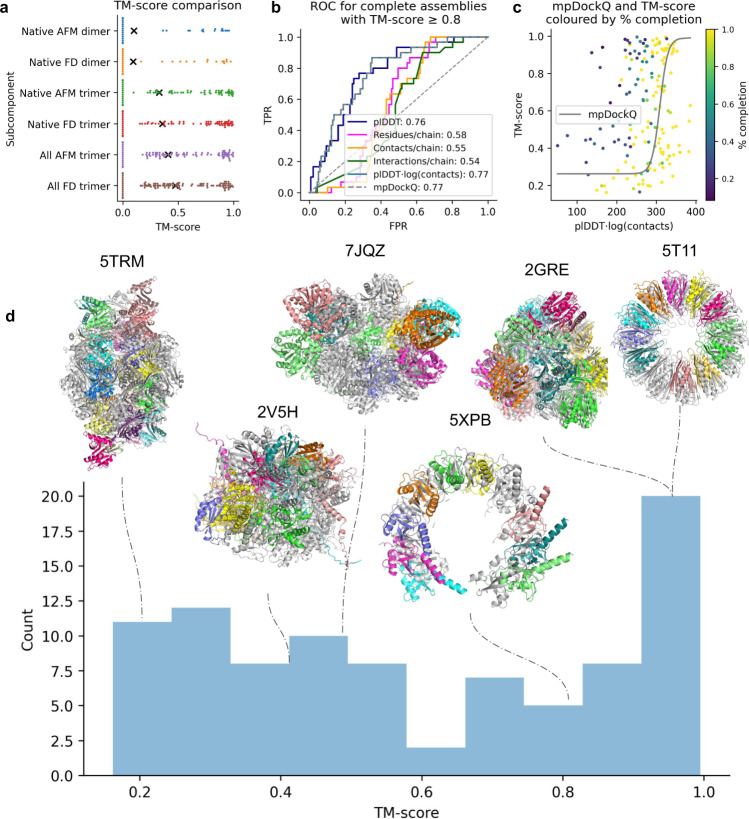
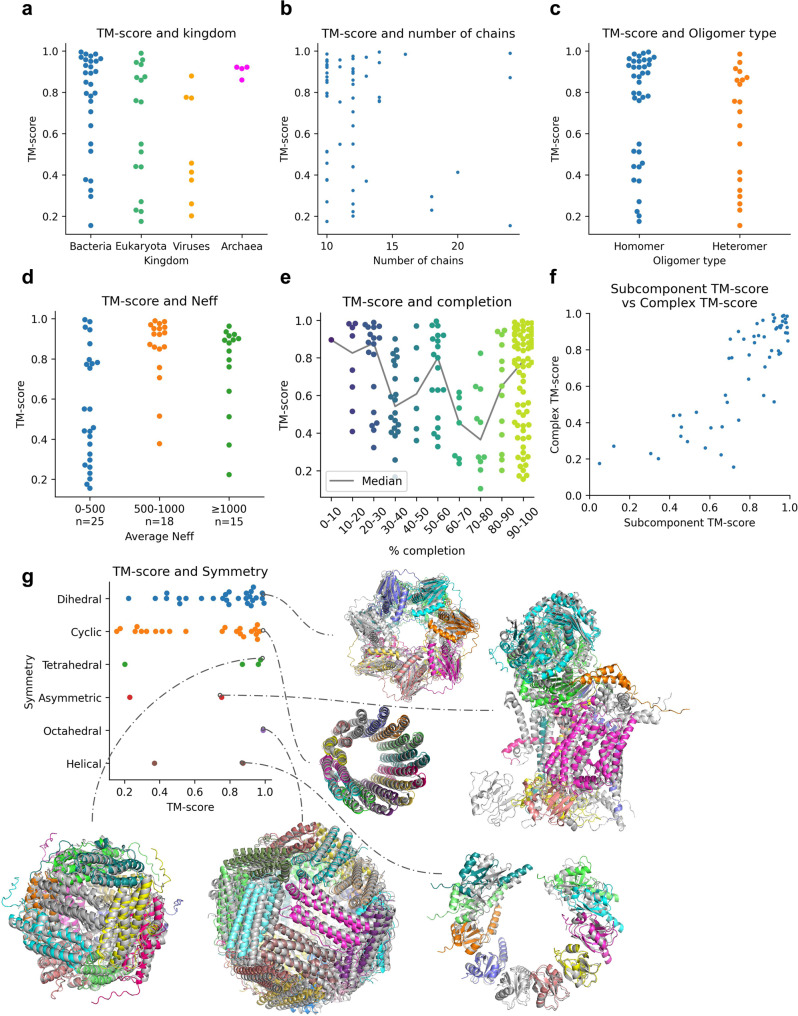
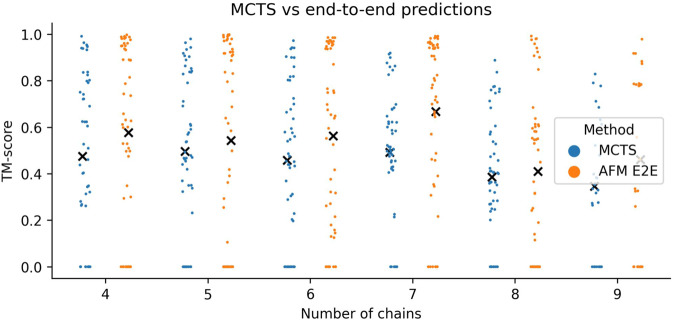
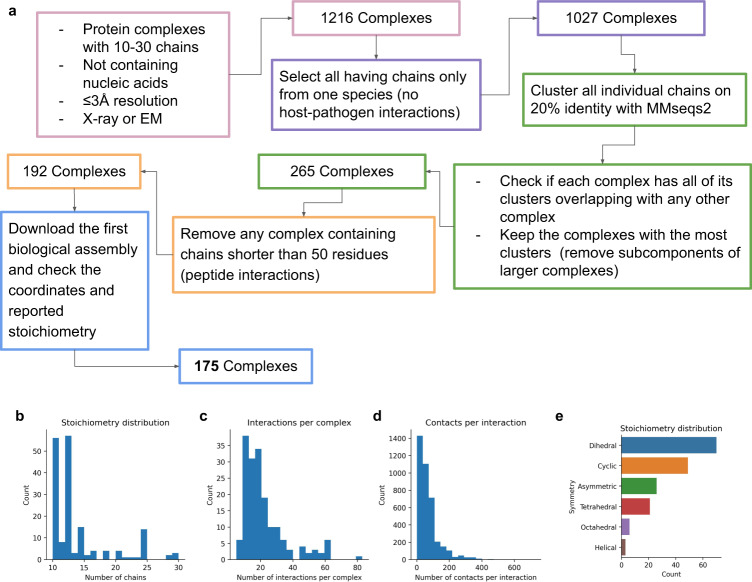
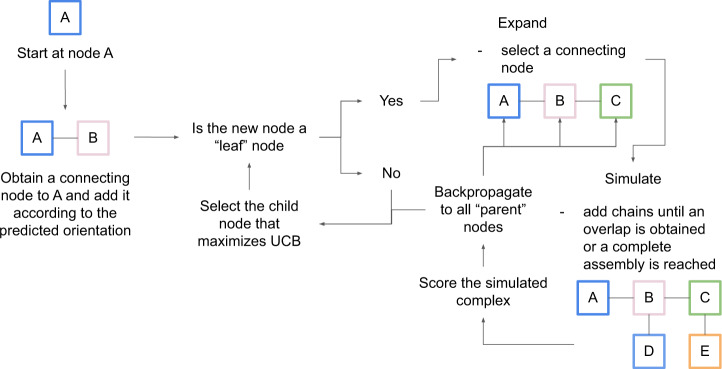
AlphaFold can predict the structure of single- and multiple-chain proteins with very high accuracy. However, the accuracy decreases with the number of chains, and the available GPU memory limits the size of protein complexes which can be predicted. Here we show that one can predict the structure of large complexes starting from predictions of subcomponents. We assemble 91 out of 175 complexes with 10-30 chains from predicted subcomponents using Monte Carlo tree search, with a median TM-score of 0.51. There are 30 highly accurate complexes (TM-score ≥0.8, 33% of complete assemblies). We create a scoring function, mpDockQ, that can distinguish if assemblies are complete and predict their accuracy. We find that complexes containing symmetry are accurately assembled, while asymmetrical complexes remain challenging. The method is freely available and accesible as a Colab notebook https://colab.research.google.com/github/patrickbryant1/MoLPC/blob/master/MoLPC.ipynb .
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
