

Calculating Binodals and Interfacial Tension of Phase-Separated Condensates from Molecular Simulations with Finite-Size Corrections
- PMID: 36227466
- PMCID: PMC9577455
- DOI: 10.1007/978-1-0716-2663-4_1
Calculating Binodals and Interfacial Tension of Phase-Separated Condensates from Molecular Simulations with Finite-Size Corrections
Abstract
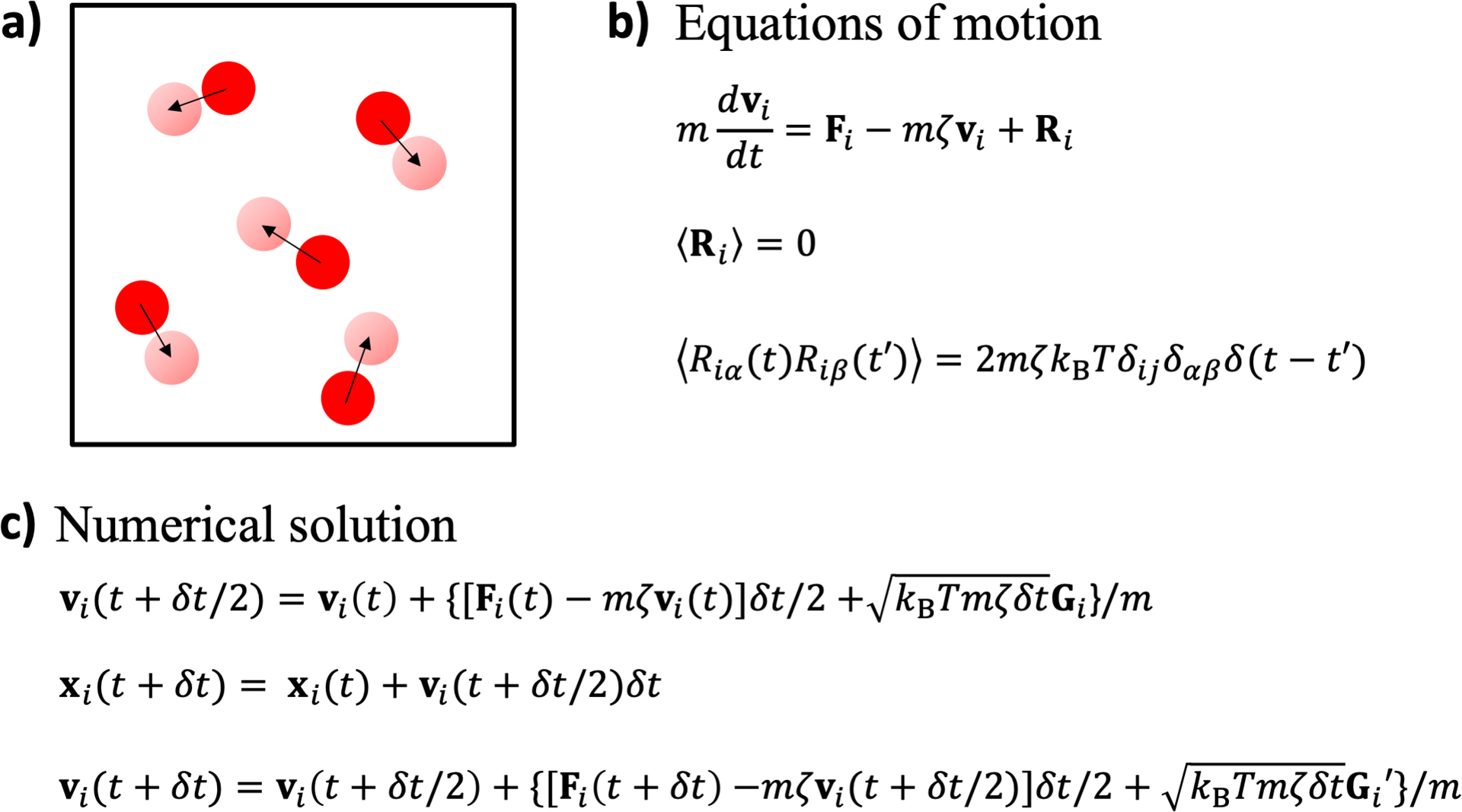
We illustrate three methods for calculating the binodals of phase-separated condensates from molecular simulations. Because molecular simulations can only be carried out for small system sizes, correction for finite sizes may be required for making direct comparison between calculated results and experimental data. We first summarize the three methods and then present detailed implementation of each method on a Lennard-Jones fluid. In the first method, chemical potentials are calculated over a range of particle densities in canonical-ensemble simulations; the densities of the dilute and dense phases at the given temperature are then found by a Maxwell equal-area construction. In Gibbs-ensemble Monte Carlo, the exchange between separated dilute and dense phases is simulated to obtain their densities. Lastly, slab-geometry molecular dynamics simulations model the dilute and dense phases in coexistence and yield not only their densities but also their interfacial tension. The three types of simulations are carried out for a range of system sizes, and the results are scaled to generate the binodals corrected for finite system sizes. Size-corrected interfacial tension is also produced from slab-geometry molecular dynamics simulations.
Keywords: Binodals; Biomolecular condensates; FMAP; Finite-size scaling; Interfacial tension; Lennard-Jones fluid; Molecular dynamics simulation; Monte-Carlo simulation; Phase separation.
© 2023. The Author(s), under exclusive license to Springer Science+Business Media, LLC, part of Springer Nature.
Figures















Similar articles
-
SpiDec: Computing binodals and interfacial tension of biomolecular condensates from simulations of spinodal decomposition.Front Mol Biosci. 2022 Oct 24;9:1021939. doi: 10.3389/fmolb.2022.1021939. eCollection 2022. Front Mol Biosci. 2022. PMID: 36353733 Free PMC article.
-
Can we approach the gas-liquid critical point using slab simulations of two coexisting phases?J Chem Phys. 2016 Sep 28;145(12):124702. doi: 10.1063/1.4962820. J Chem Phys. 2016. PMID: 27782674
-
The interfacial tension and phase diagram of the Widom-Rowlinson mixture via Monte Carlo simulations.J Chem Phys. 2008 Jan 7;128(1):014712. doi: 10.1063/1.2806279. J Chem Phys. 2008. PMID: 18190217
-
Accurate and precise determination of critical properties from Gibbs ensemble Monte Carlo simulations.J Chem Phys. 2015 Sep 21;143(11):114113. doi: 10.1063/1.4930848. J Chem Phys. 2015. PMID: 26395693
-
Fluctuations, Finite-Size Effects and the Thermodynamic Limit in Computer Simulations: Revisiting the Spatial Block Analysis Method.Entropy (Basel). 2018 Mar 24;20(4):222. doi: 10.3390/e20040222. Entropy (Basel). 2018. PMID: 33265313 Free PMC article. Review.
Cited by
-
Fundamental Aspects of Phase-Separated Biomolecular Condensates.Chem Rev. 2024 Jul 10;124(13):8550-8595. doi: 10.1021/acs.chemrev.4c00138. Epub 2024 Jun 17. Chem Rev. 2024. PMID: 38885177 Free PMC article. Review.
-
Atomistic Modeling of Liquid-Liquid Phase Equilibrium Explains Dependence of Critical Temperature on γ-Crystallin Sequence.bioRxiv [Preprint]. 2023 Apr 28:2023.04.25.538329. doi: 10.1101/2023.04.25.538329. bioRxiv. 2023. Update in: Commun Biol. 2023 Aug 29;6(1):886. doi: 10.1038/s42003-023-05270-7. PMID: 37162827 Free PMC article. Updated. Preprint.
-
Atomistic modeling of liquid-liquid phase equilibrium explains dependence of critical temperature on γ-crystallin sequence.Commun Biol. 2023 Aug 29;6(1):886. doi: 10.1038/s42003-023-05270-7. Commun Biol. 2023. PMID: 37644195 Free PMC article.
-
SpiDec: Computing binodals and interfacial tension of biomolecular condensates from simulations of spinodal decomposition.Front Mol Biosci. 2022 Oct 24;9:1021939. doi: 10.3389/fmolb.2022.1021939. eCollection 2022. Front Mol Biosci. 2022. PMID: 36353733 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
