

Loss of non-motor kinesin KIF26A causes congenital brain malformations via dysregulated neuronal migration and axonal growth as well as apoptosis
- PMID: 36228617
- PMCID: PMC10585591
- DOI: 10.1016/j.devcel.2022.09.011
Loss of non-motor kinesin KIF26A causes congenital brain malformations via dysregulated neuronal migration and axonal growth as well as apoptosis
Abstract
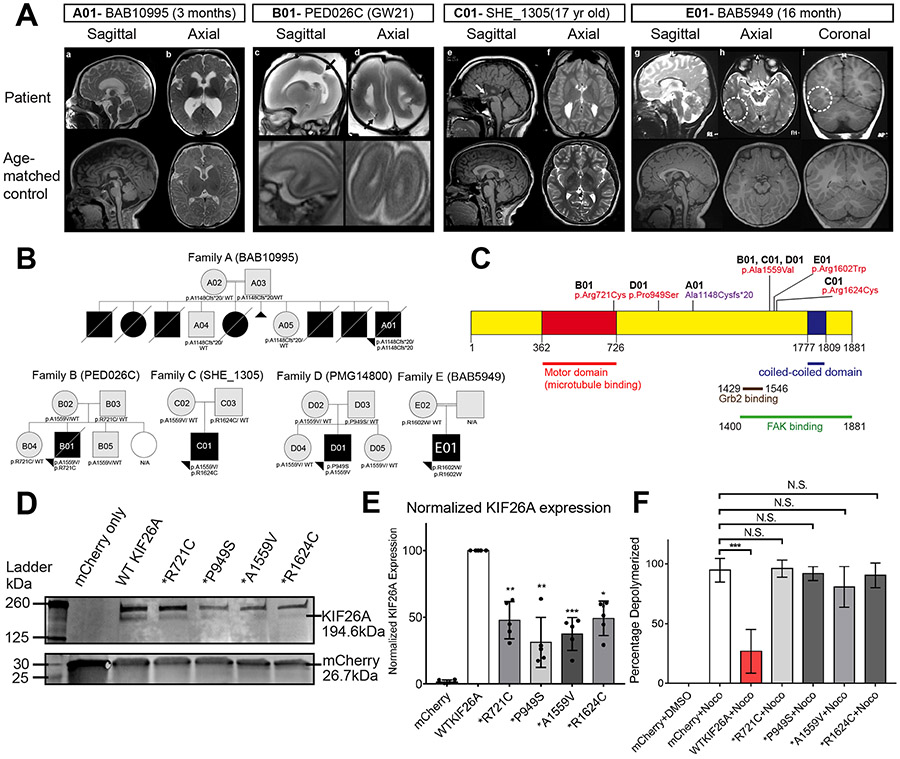
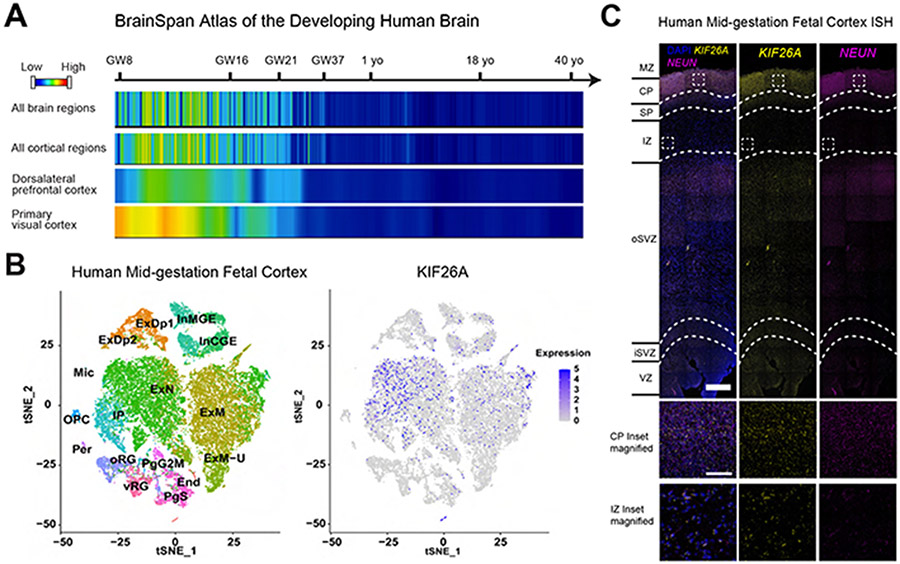
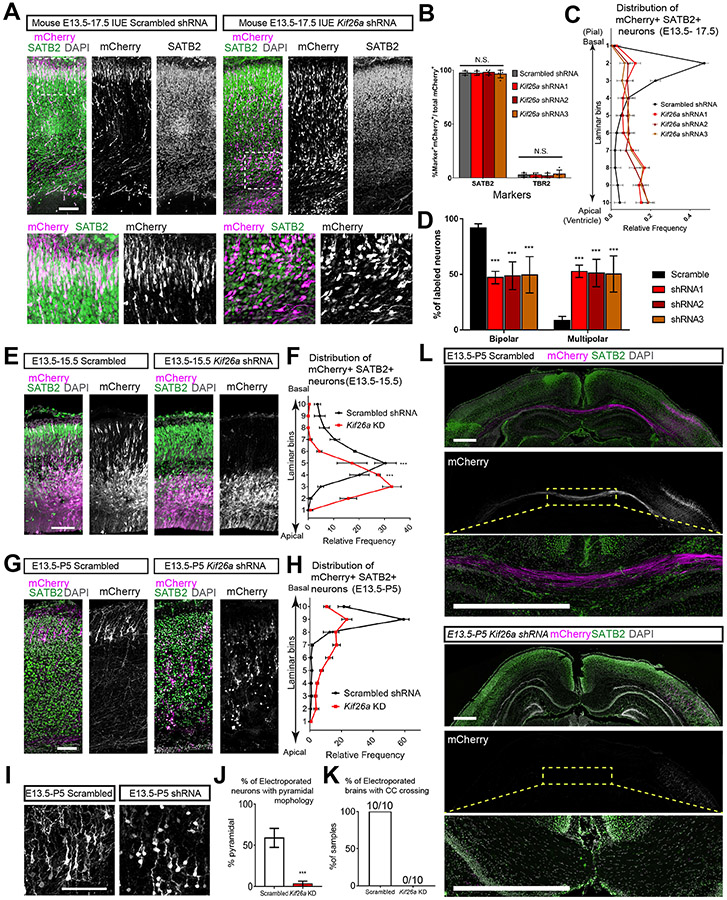
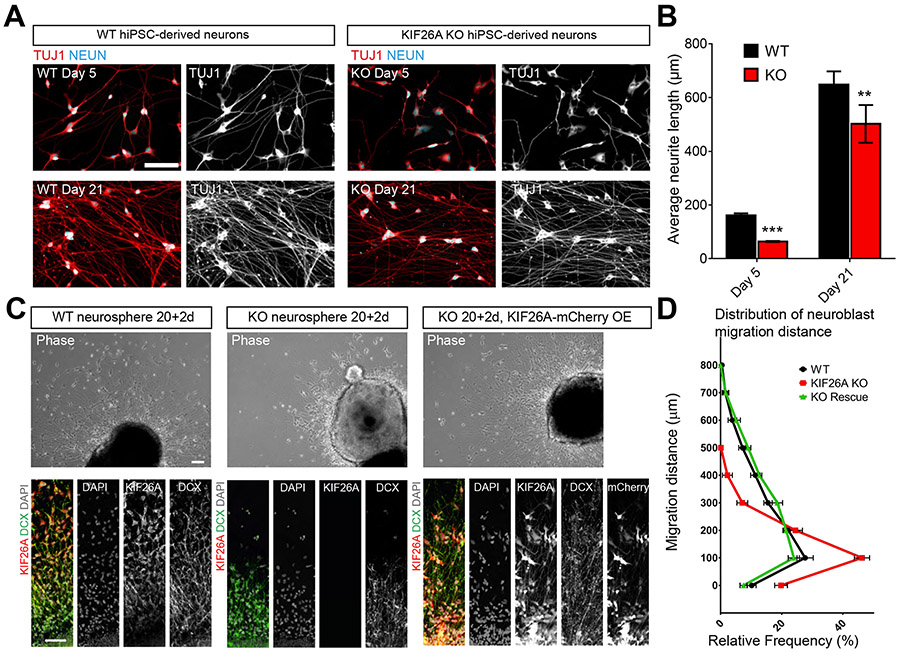
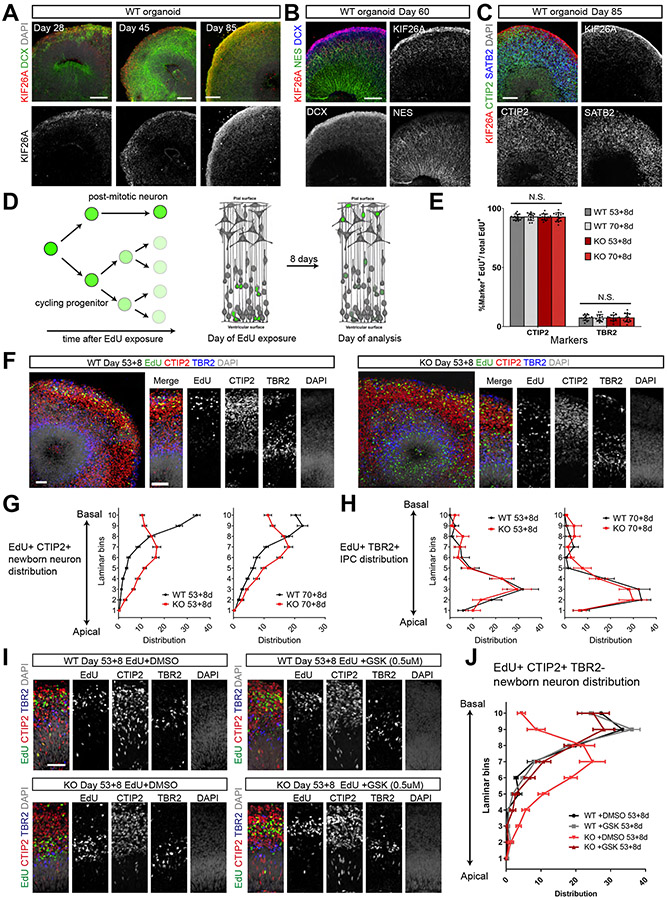
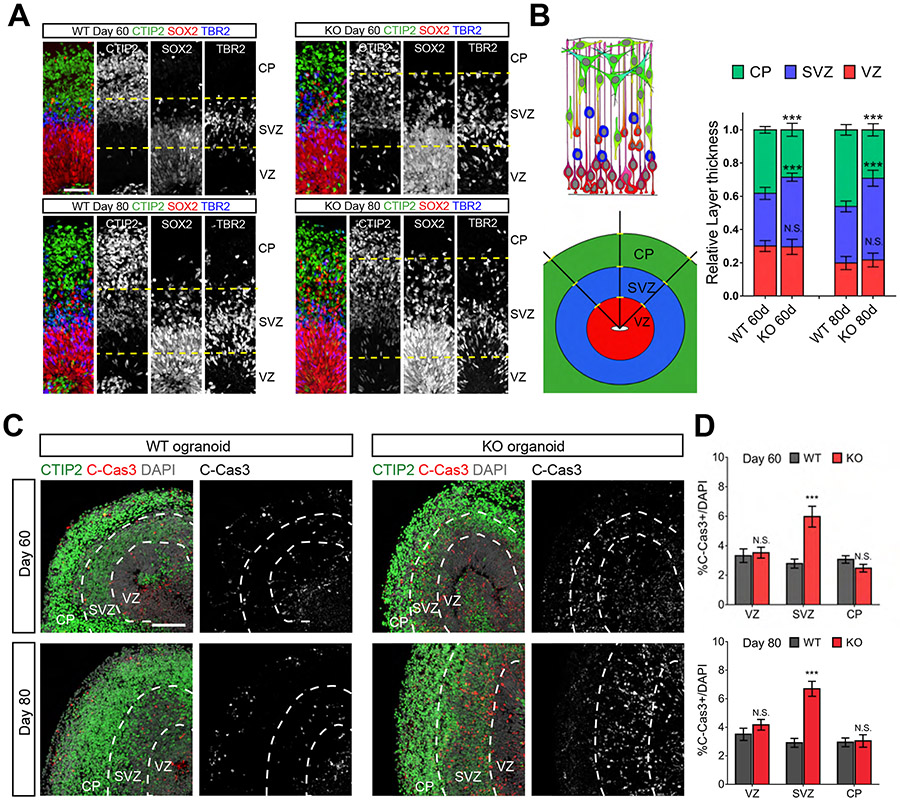
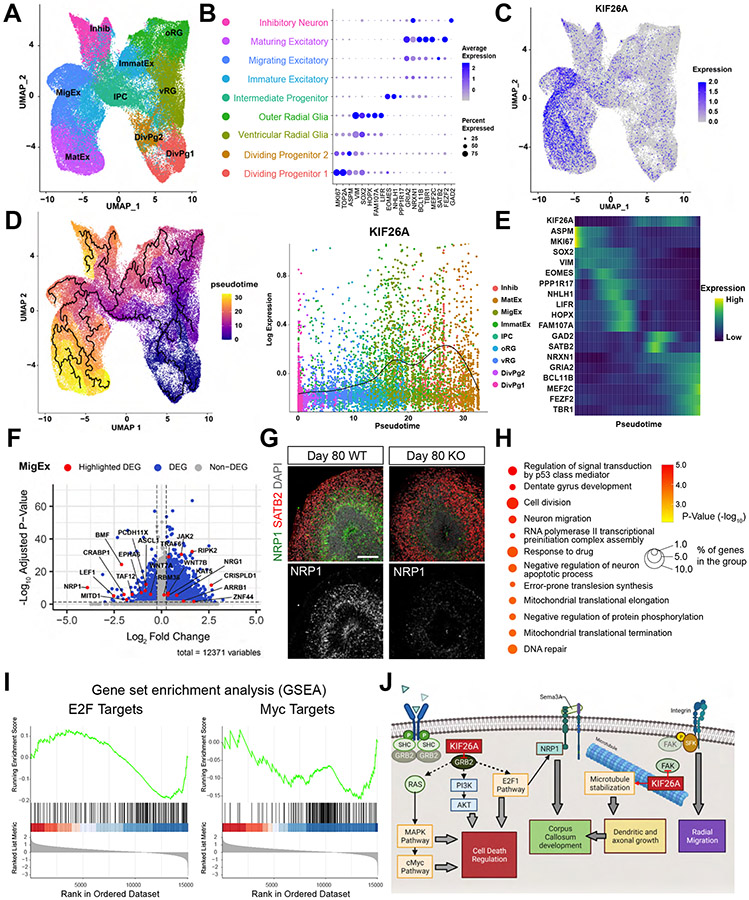
Kinesins are canonical molecular motors but can also function as modulators of intracellular signaling. KIF26A, an unconventional kinesin that lacks motor activity, inhibits growth-factor-receptor-bound protein 2 (GRB2)- and focal adhesion kinase (FAK)-dependent signal transduction, but its functions in the brain have not been characterized. We report a patient cohort with biallelic loss-of-function variants in KIF26A, exhibiting a spectrum of congenital brain malformations. In the developing brain, KIF26A is preferentially expressed during early- and mid-gestation in excitatory neurons. Combining mice and human iPSC-derived organoid models, we discovered that loss of KIF26A causes excitatory neuron-specific defects in radial migration, localization, dendritic and axonal growth, and apoptosis, offering a convincing explanation of the disease etiology in patients. Single-cell RNA sequencing in KIF26A knockout organoids revealed transcriptional changes in MAPK, MYC, and E2F pathways. Our findings illustrate the pathogenesis of KIF26A loss-of-function variants and identify the surprising versatility of this non-motor kinesin.
Keywords: apoptosis; cerebral cortex; congenital brain malformation; corpus callosum; development; genetics; kinesin; migration; organoid.
Copyright © 2022 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests J.R.L. has stock ownership in 23andMe, is a paid consultant for the Regeneron Genetics Center, and is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting. The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing conducted at Baylor Genetics (BG) Laboratories, and J.R.L. is a member of the Scientific Advisory Board of BG. C.A.W. has stock ownership in Maze Therapeutics and is a paid consultant for Third Rock Ventures and Flagship Pioneering.
Figures
References
-
- Asselin L, Rivera Alvarez J, Heide S, Bonnet CS, Tilly P, Vitet H, Weber C, Bacino CA, Baranano K, Chassevent A, et al. (2020). Mutations in the KIF21B kinesin gene cause neurodevelopmental disorders through imbalanced canonical motor activity. Nat Commun 11, 2441 DOI: 10.1038/s41467-020-16294-6. - DOI - PMC - PubMed
-
- Bershteyn M, Nowakowski TJ, Pollen AA, Di Lullo E, Nene A, Wynshaw-Boris A, and Kriegstein AR (2017). Human iPSC-Derived Cerebral Organoids Model Cellular Features of Lissencephaly and Reveal Prolonged Mitosis of Outer Radial Glia. Cell Stem Cell 20, 435–449 e434 DOI: 10.1016/j.stem.2016.12.007. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 NS032457/NS/NINDS NIH HHS/United States
- UM1 HG008900/HG/NHGRI NIH HHS/United States
- R01 HG009141/HG/NHGRI NIH HHS/United States
- R35 NS105078/NS/NINDS NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- T32 GM144273/GM/NIGMS NIH HHS/United States
- R01 NS035129/NS/NINDS NIH HHS/United States
- U54 HD090255/HD/NICHD NIH HHS/United States
- R01 GM106373/GM/NIGMS NIH HHS/United States
- R25 NS070682/NS/NINDS NIH HHS/United States
- U01 HG011758/HG/NHGRI NIH HHS/United States
- T32 GM007753/GM/NIGMS NIH HHS/United States
- UM1 HG006542/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
