

High-resolution structure of a fish aquaporin reveals a novel extracellular fold
- PMID: 36229063
- PMCID: PMC9559756
- DOI: 10.26508/lsa.202201491
High-resolution structure of a fish aquaporin reveals a novel extracellular fold
Abstract
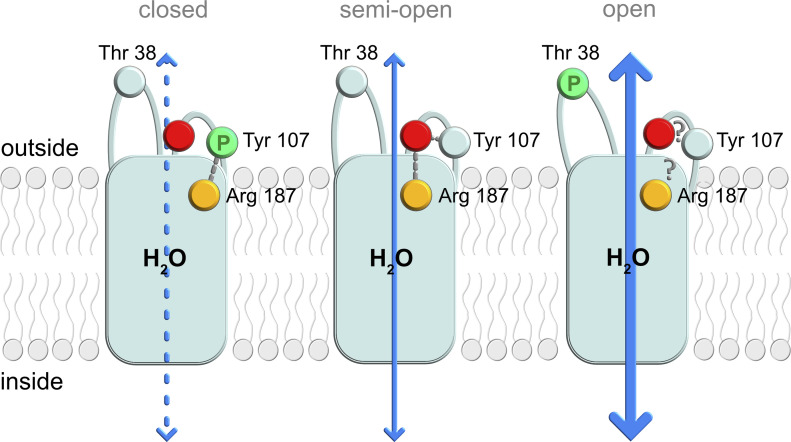
Aquaporins are protein channels embedded in the lipid bilayer in cells from all organisms on earth that are crucial for water homeostasis. In fish, aquaporins are believed to be important for osmoregulation; however, the molecular mechanism behind this is poorly understood. Here, we present the first structural and functional characterization of a fish aquaporin; cpAQP1aa from the fresh water fish climbing perch (<i>Anabas testudineus</i>), a species that is of high osmoregulatory interest because of its ability to spend time in seawater and on land. These studies show that cpAQP1aa is a water-specific aquaporin with a unique fold on the extracellular side that results in a constriction region. Functional analysis combined with molecular dynamic simulations suggests that phosphorylation at two sites causes structural perturbations in this region that may have implications for channel gating from the extracellular side.
© 2022 Zeng et al.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures














References
-
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 68: 352–367. 10.1107/s0907444912001308 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Miscellaneous