

Overexpression of mouse prion protein in transgenic mice causes a non-transmissible spongiform encephalopathy
- PMID: 36229637
- PMCID: PMC9562354
- DOI: 10.1038/s41598-022-21608-3
Overexpression of mouse prion protein in transgenic mice causes a non-transmissible spongiform encephalopathy
Abstract
Transgenic mice over-expressing human PRNP or murine Prnp transgenes on a mouse prion protein knockout background have made key contributions to the understanding of human prion diseases and have provided the basis for many of the fundamental advances in prion biology, including the first report of synthetic mammalian prions. In this regard, the prion paradigm is increasingly guiding the exploration of seeded protein misfolding in the pathogenesis of other neurodegenerative diseases. Here we report that a well-established and widely used line of such mice (Tg20 or tga20), which overexpress wild-type mouse prion protein, exhibit spontaneous aggregation and accumulation of misfolded prion protein in a strongly age-dependent manner, which is accompanied by focal spongiosis and occasional neuronal loss. In some cases a clinical syndrome developed with phenotypic features that closely resemble those seen in prion disease. However, passage of brain homogenate from affected, aged mice failed to transmit this syndrome when inoculated intracerebrally into further recipient animals. We conclude that overexpression of the wild-type mouse prion protein can cause an age-dependent protein misfolding disorder or proteinopathy that is not associated with the production of an infectious agent but can produce a phenotype closely similar to authentic prion disease.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no conflicts of interest relevant to this manuscript, with the exceptions of: J.C. is a Director and G.S.J., J.D.F.W. and J.C. are shareholders of D-Gen Limited (London), an academic spin-out company working in the field of prion disease diagnosis, decontamination and therapeutics. D-Gen supplied the antibody ICSM35 used in this study.
Figures
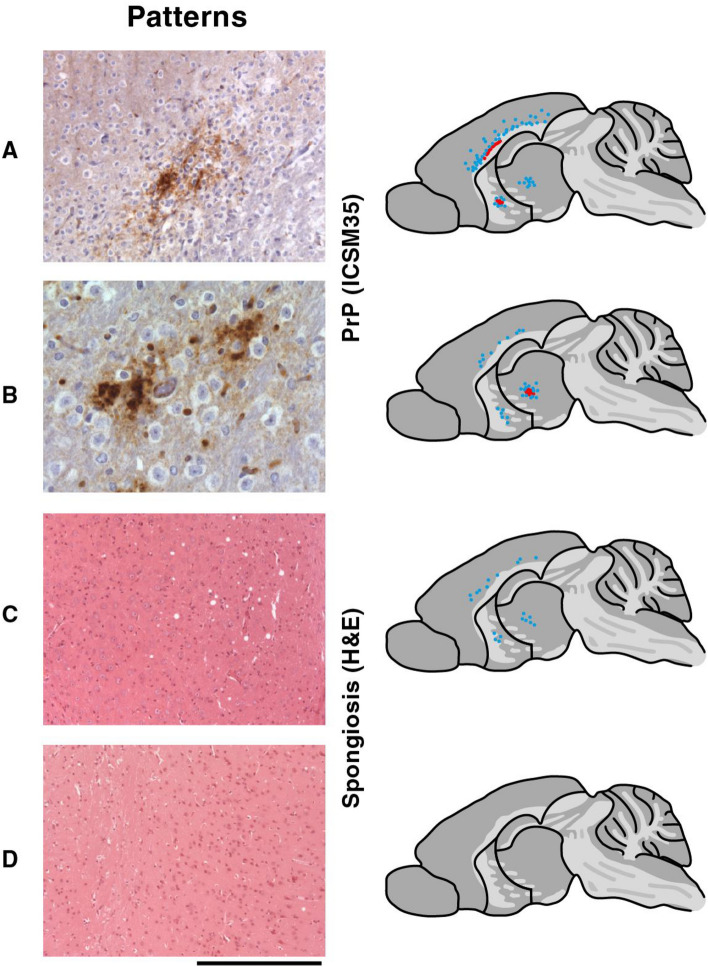

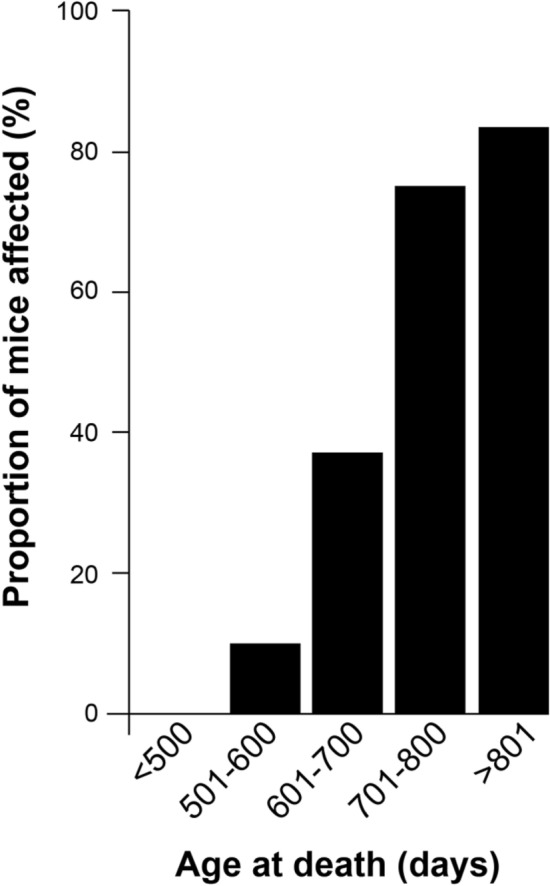
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
