

Circulating Tumor DNA: Less Invasive, More Representative Method to Unveil the Genomic Landscape of Newly Diagnosed Multiple Myeloma Than Bone Marrow Aspirates
- PMID: 36230837
- PMCID: PMC9563595
- DOI: 10.3390/cancers14194914
Circulating Tumor DNA: Less Invasive, More Representative Method to Unveil the Genomic Landscape of Newly Diagnosed Multiple Myeloma Than Bone Marrow Aspirates
Abstract
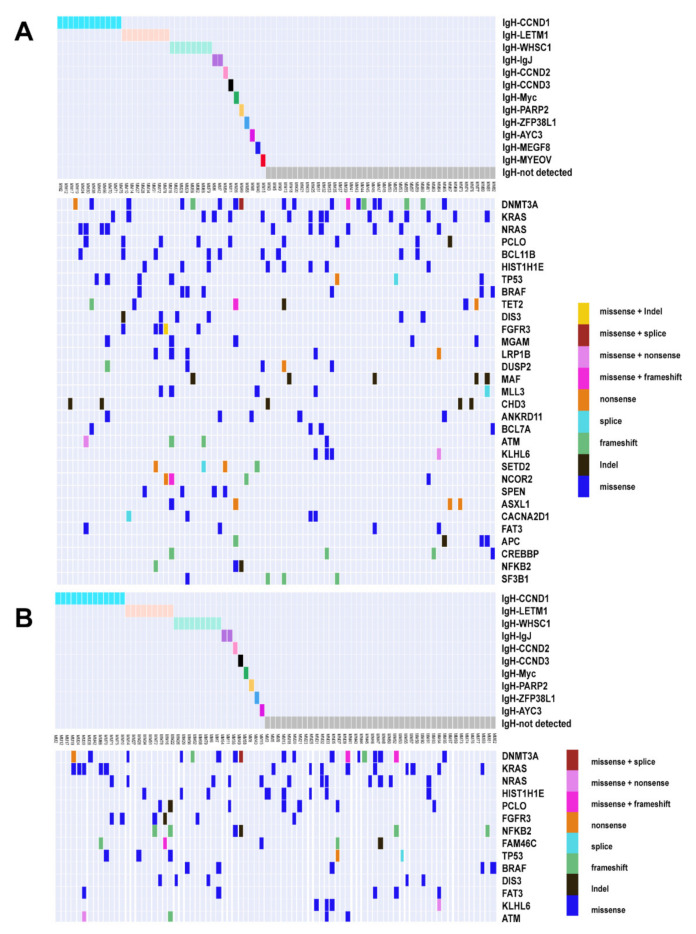
Multiple myeloma (MM) is highly heterogenous and dynamic in its genomic abnormalities. Capturing a representative image of these alterations is essential in understanding the molecular pathogenesis and progression of the disease but was limited by single-site invasive bone marrow (BM) biopsy-based genomics studies. We compared the mutational landscapes of circulating tumor DNA (ctDNA) and BM in 82 patients with newly diagnosed MM. A 413-gene panel was used in the sequencing. Our results showed that more than 70% of MM patients showed one or more genes with somatic mutations and at least half of the mutated genes were shared between ctDNA and BM samples. Compared to the BM samples, ctDNA exhibited more types of driver mutations in the shared driver genes, higher numbers of uniquely mutated genes and subclonal clusters, more translocation-associated mutations, and higher frequencies of mutated genes enriched in the transcriptional regulation pathway. Multivariate Cox analysis showed that age, ctDNA mutations in the transcriptional regulation pathway and DNA repair pathway were independent predictors of progression-free survival (PFS). Our results demonstrated sequencing of ctDNA provides more thorough information on the genomic instability and is a potential representative biomarker for risk stratification and in newly diagnosed MM than bone marrow.
Keywords: circulating tumor DNA; multiple myeloma; prognosis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures







References
-
- Gertz M.A., Lacy M.Q., Dispenzieri A., Greipp P.R., Litzow M.R., Henderson K.J., Van Wier S.A., Ahmann G.J., Fonseca R. Clinical implications of t(11;14)(q13;q32), t(4;14)(p16.3;q32), and -17p13 in myeloma patients treated with high-dose therapy. Blood. 2005;106:2837–2840. doi: 10.1182/blood-2005-04-1411. - DOI - PMC - PubMed
-
- Lohr J.G., Stojanov P., Carter S.L., Cruz-Gordillo P., Lawrence M.S., Auclair D., Sougnez C., Knoechel B., Gould J., Saksena G., et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell. 2014;25:91–101. doi: 10.1016/j.ccr.2013.12.015. - DOI - PMC - PubMed
-
- Walker B.A., Mavrommatis K., Wardell C.P., Ashby T.C., Bauer M., Davies F.E., Rosenthal A., Wang H., Qu P., Hoering A., et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood. 2018;132:587–597. doi: 10.1182/blood-2018-03-840132. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
