

AMBRA1 p.Gln30Arg Mutation, Identified in a Cowden Syndrome Family, Exhibits Hyperproliferative Potential in hTERT-RPE1 Cells
- PMID: 36232425
- PMCID: PMC9570079
- DOI: 10.3390/ijms231911124
AMBRA1 p.Gln30Arg Mutation, Identified in a Cowden Syndrome Family, Exhibits Hyperproliferative Potential in hTERT-RPE1 Cells
Abstract
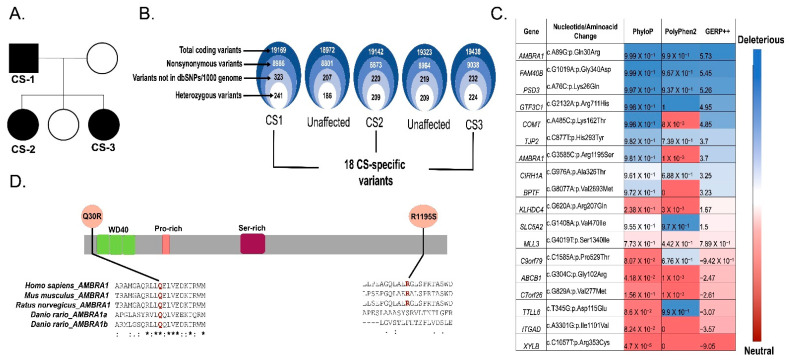
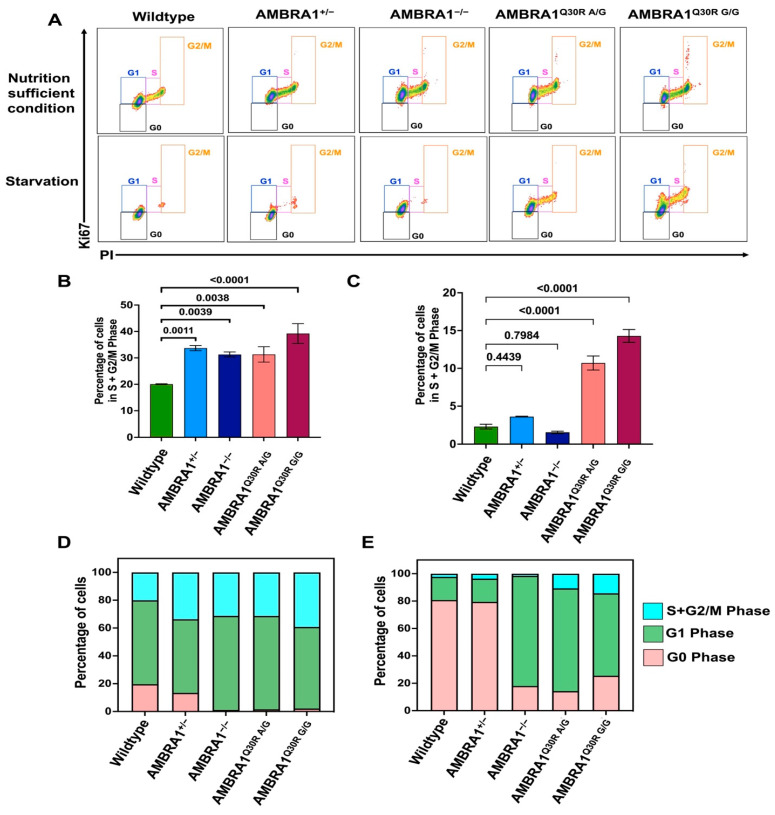
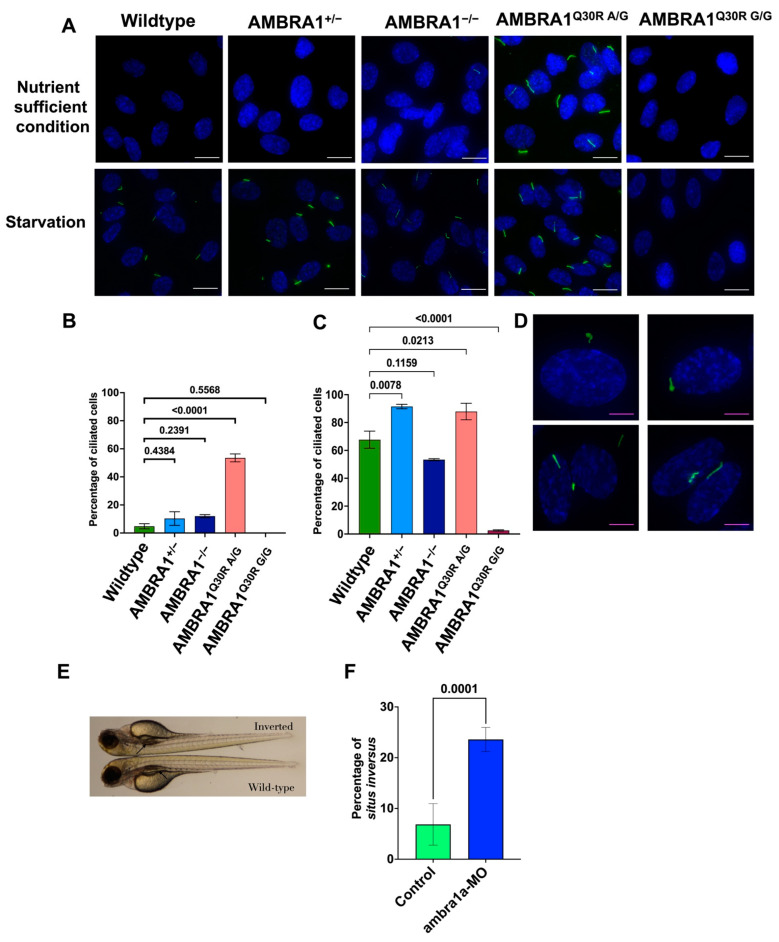
Cowden syndrome (CS) is a rare autosomal dominant disorder associated with multiple hamartomatous and neoplastic lesions in various organs. Most CS patients have been found to have germline mutations in the PTEN tumor suppressor. In the present study, we investigated the causative gene of CS in a family of PTEN (phosphatase and tensin homolog deleted on chromosome 10) -negative CS patients. Whole exome sequencing analysis revealed AMBRA1 (Autophagy and Beclin 1 Regulator 1) as a novel candidate gene harboring two germline variants: p.Gln30Arg (Q30R) and p.Arg1195Ser (R1195S). AMBRA1 is a key regulator of the autophagy signaling network and a tumor suppressor. To functionally validate the role of AMBRA1 in the clinical manifestations of CS, we generated AMBRA1 depletion and Q30R mutation in hTERT-RPE1 (humanTelomerase Reverse Transcriptase-immortalized Retinal Pigmented Epithelial cells) using the CRISPR-Cas9 gene editing system. We observed that both AMBRA1-depleted and mutant cells showed accumulation in the S phase, leading to hyperproliferation, which is a characteristic of hamartomatous lesions. Specifically, the AMBRA1 Q30R mutation disturbed the G1/S transition of cells, leading to continuous mitotic entry of mutant cells, irrespective of the extracellular condition. From our analysis of primary ciliogenesis in these cells, we speculated that the mitotic entry of AMBRA1 Q30R mutants could be due to non-functional primary cilia that lead to impaired processing of extracellular sensory signals. Additionally, we observed a situs inversus phenotype in ambra1-depleted zebrafish, a developmental abnormality resulting from dysregulated primary ciliogenesis. Taken together, we established that the AMBRA1 Q30R mutation that we observed in CS patients might play an important role in inducing the hyperproliferative potential of cells through regulating primary ciliogenesis.
Keywords: AMBRA1; AMBRA1 Q30R mutant; CRISPR/Cas; Cowden syndrome; G1/S transition; primary cilia.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Identification of a Cowden syndrome patient with a novel PTEN mutation and establishment of patient-derived induced pluripotent stem cells.In Vitro Cell Dev Biol Anim. 2022 Jan;58(1):69-78. doi: 10.1007/s11626-021-00637-8. Epub 2022 Jan 3. In Vitro Cell Dev Biol Anim. 2022. PMID: 34984555 Free PMC article.
-
Novel Germline PTEN Mutation Associated with Cowden Syndrome and Osteosarcoma.Cancer Genomics Proteomics. 2018 Mar-Apr;15(2):115-120. doi: 10.21873/cgp.20069. Cancer Genomics Proteomics. 2018. PMID: 29496690 Free PMC article.
-
Germline epigenetic regulation of KILLIN in Cowden and Cowden-like syndrome.JAMA. 2010 Dec 22;304(24):2724-31. doi: 10.1001/jama.2010.1877. JAMA. 2010. PMID: 21177507 Free PMC article.
-
Breast cancer risk and clinical implications for germline PTEN mutation carriers.Breast Cancer Res Treat. 2017 Aug;165(1):1-8. doi: 10.1007/s10549-015-3665-z. Epub 2015 Dec 23. Breast Cancer Res Treat. 2017. PMID: 26700035 Review.
-
Cowden syndrome: a critical review of the clinical literature.J Genet Couns. 2009 Feb;18(1):13-27. doi: 10.1007/s10897-008-9187-7. Epub 2008 Oct 30. J Genet Couns. 2009. PMID: 18972196 Review.
Cited by
-
Molecular and genetic evidence for the role of AMBRA1 in suppressing S-phase entry and tumorigenesis.iScience. 2025 Jul 5;28(8):113054. doi: 10.1016/j.isci.2025.113054. eCollection 2025 Aug 15. iScience. 2025. PMID: 40734673 Free PMC article.
-
Uncommon but Important: Tertiary Center Experience with Rare Cases of Breast Hamartoma.Life (Basel). 2025 Jul 5;15(7):1076. doi: 10.3390/life15071076. Life (Basel). 2025. PMID: 40724578 Free PMC article.
References
-
- Garofola C., Jamal Z., Gross G.P. Cowden Disease. StatPearls Publishing; Treasure Island, FL, USA: 2021. - PubMed
-
- Yehia L., Ni Y., Sesock K., Niazi F., Fletcher B., Chen H.J.L., LaFramboise T., Eng C. Unexpected cancer-predisposition gene variants in Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome patients without underlying germline PTEN mutations. PLoS Genet. 2018;14:e1007352. doi: 10.1371/journal.pgen.1007352. - DOI - PMC - PubMed
-
- Marsh D.J., Coulon V., Lunetta K.L., Rocca-Serra P., Dahia P.L., Zheng Z., Liaw D., Caron S., Duboué B., Lin A.Y., et al. Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum. Mol. Genet. 1998;7:507–515. doi: 10.1093/hmg/7.3.507. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
