

Molecular Features of SLC26A4 Common Variant p.L117F
- PMID: 36233414
- PMCID: PMC9570580
- DOI: 10.3390/jcm11195549
Molecular Features of SLC26A4 Common Variant p.L117F
Abstract
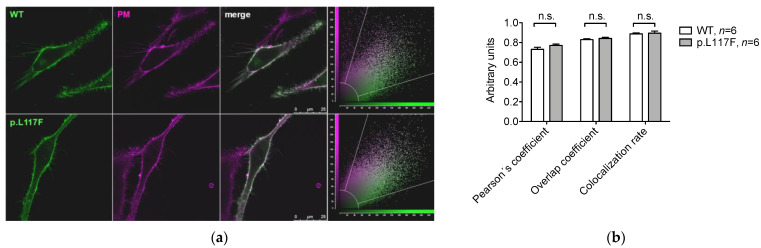
The SLC26A4 gene, which encodes the anion exchanger pendrin, is involved in determining syndromic (Pendred syndrome) and non-syndromic (DFNB4) autosomal recessive hearing loss. SLC26A4 c.349C>T, p.L117F is a relatively common allele in the Ashkenazi Jewish community, where its minor allele frequency is increased compared to other populations. Although segregation and allelic data support the pathogenicity of this variant, former functional tests showed characteristics that were indistinguishable from those of the wild-type protein. Here, we applied a triad of cell-based assays, i.e., measurement of the ion transport activity by a fluorometric method, determination of the subcellular localization by confocal microscopy, and assessment of protein expression levels, to conclusively assign or exclude the pathogenicity of SLC26A4 p.L117F. This protein variant showed a moderate, but significant, reduction in ion transport function, a partial retention in the endoplasmic reticulum, and a strong reduction in expression levels as a consequence of an accelerated degradation by the Ubiquitin Proteasome System, all supporting pathogenicity. The functional and molecular features of human pendrin p.L117F were recapitulated by the mouse ortholog, thus indicating that a mouse carrying this variant might represent a good model of Pendred syndrome/DFNB4.
Keywords: DFNB4; Pendred syndrome; SLC26A4; functional testing; hearing loss; pendrin.
Conflict of interest statement
The authors declare no conflict of interest. The funder had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Figures






Similar articles
-
Inhibitors of the ubiquitin‑proteasome system rescue cellular levels and ion transport function of pathogenic pendrin (SLC26A4) protein variants.Int J Mol Med. 2025 May;55(5):69. doi: 10.3892/ijmm.2025.5510. Epub 2025 Mar 7. Int J Mol Med. 2025. PMID: 40052591 Free PMC article.
-
Novel small molecule-mediated restoration of the surface expression and anion exchange activity of mutated pendrin causing Pendred syndrome and DFNB4.Biomed Pharmacother. 2023 Nov;167:115445. doi: 10.1016/j.biopha.2023.115445. Epub 2023 Sep 9. Biomed Pharmacother. 2023. PMID: 37690388
-
Identification of novel functional null allele of SLC26A4 associated with enlarged vestibular aqueduct and its possible implication.Audiol Neurootol. 2014;19(5):319-26. doi: 10.1159/000366190. Epub 2014 Oct 24. Audiol Neurootol. 2014. PMID: 25358692
-
Genetic architecture and phenotypic landscape of SLC26A4-related hearing loss.Hum Genet. 2022 Apr;141(3-4):455-464. doi: 10.1007/s00439-021-02311-1. Epub 2021 Aug 3. Hum Genet. 2022. PMID: 34345941 Review.
-
Molecular and functional characterization of human pendrin and its allelic variants.Cell Physiol Biochem. 2011;28(3):451-66. doi: 10.1159/000335107. Epub 2011 Nov 18. Cell Physiol Biochem. 2011. PMID: 22116358 Review.
Cited by
-
Novel genetic determinants contribute to hearing loss in a central European cohort with enlarged vestibular aqueduct.Mol Med. 2025 Mar 22;31(1):111. doi: 10.1186/s10020-025-01159-9. Mol Med. 2025. PMID: 40121402 Free PMC article.
-
SLC26A4 C.317C > A Variant: Functional Analysis and Patient-Derived Induced Pluripotent Stem Line Development.Mol Genet Genomic Med. 2025 Apr;13(4):e70098. doi: 10.1002/mgg3.70098. Mol Genet Genomic Med. 2025. PMID: 40260864 Free PMC article.
References
-
- Royaux I.E., Wall S.M., Karniski L.P., Everett L.A., Suzuki K., Knepper M.A., Green E.D. Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc. Natl. Acad. Sci. USA. 2001;98:4221–4226. doi: 10.1073/pnas.071516798. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
