

Evaluating Genetic Modifiers of Duchenne Muscular Dystrophy Disease Progression Using Modeling and MRI
- PMID: 36240102
- PMCID: PMC9687406
- DOI: 10.1212/WNL.0000000000201163
Evaluating Genetic Modifiers of Duchenne Muscular Dystrophy Disease Progression Using Modeling and MRI
Abstract
Background and objectives: Duchenne muscular dystrophy (DMD) is a progressive muscle degenerative disorder with a well-characterized disease phenotype but considerable interindividual heterogeneity that is not well understood. The aim of this study was to evaluate the effects of dystrophin variations and genetic modifiers of DMD on rate and age of muscle replacement by fat.
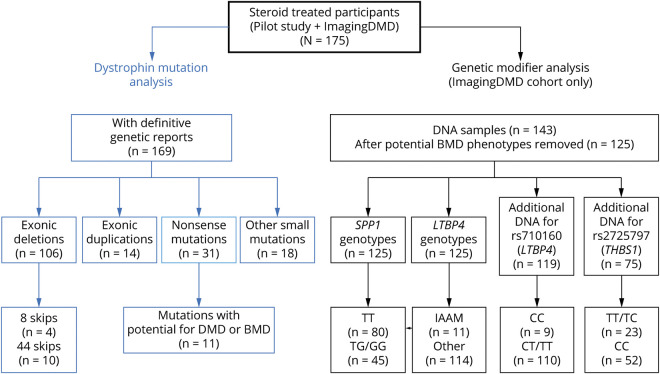
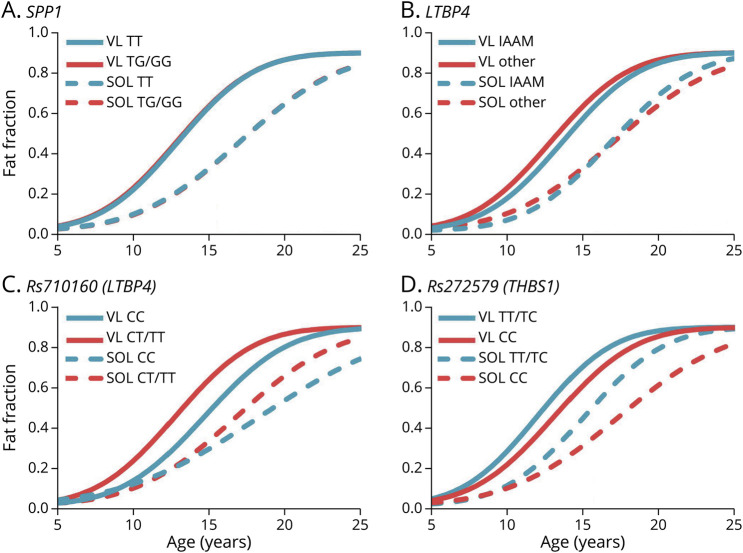
Methods: One hundred seventy-five corticosteroid treated participants from the ImagingDMD natural history study underwent repeated magnetic resonance spectroscopy (MRS) of the vastus lateralis (VL) and soleus (SOL) to determine muscle fat fraction (FF). MRS was performed annually in most instances; however, some individuals had additional visits at 3 or 6 monthss intervals. FF changes over time were modeled using nonlinear mixed effects to estimate disease trajectories based on the age that the VL or SOL reached half-maximum change in FF (mu) and the time required for FF change (sigma). Computed mu and sigma values were evaluated for dystrophin variations that have demonstrated the ability to lead to a mild phenotype as well as compared between different genetic polymorphism groups.
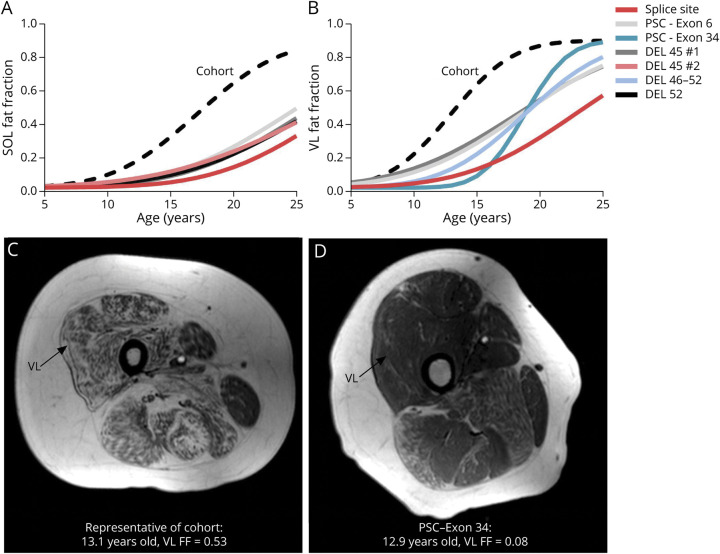
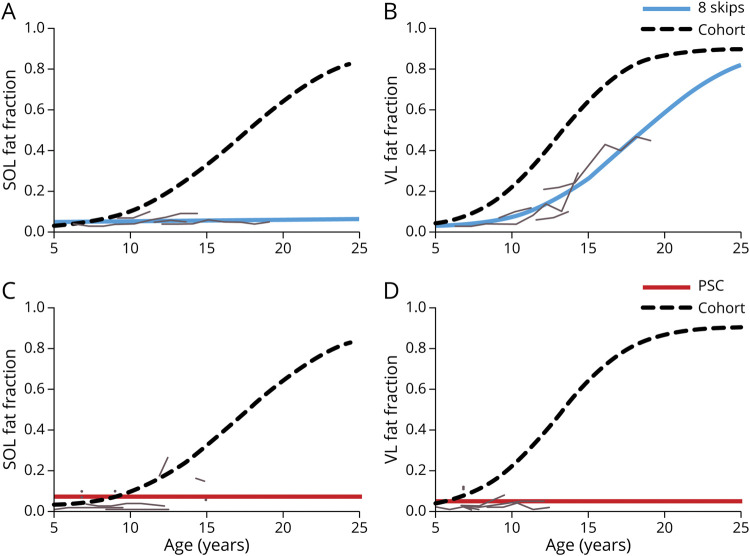
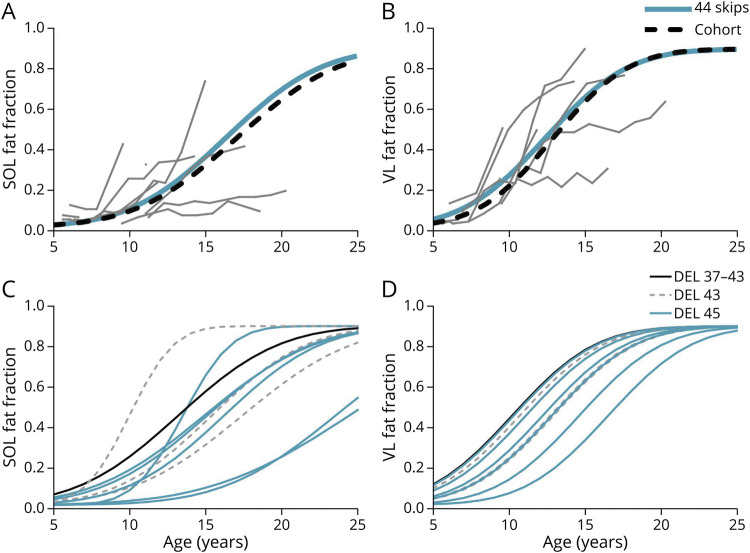
Results: Participants with dystrophin gene deletions amenable to exon 8 skipping (n = 4) had minimal increases in SOL FF and had an increase in VL mu value by 4.4 years compared with a reference cohort (p = 0.039). Participants with nonsense variations within exons that may produce milder phenotypes (n = 11) also had minimal increases in SOL and VL FFs. No differences in estimated FF trajectories were seen for individuals amenable to exon 44 skipping (n = 10). Modeling of the SPP1, LTBP4, and thrombospondin-1 (THBS1) genetic modifiers did not result in significant differences in muscle FF trajectories between genotype groups (p > 0.05); however, trends were noted for the polymorphisms associated with long-range regulation of LTBP4 and THBS1 that deserve further follow-up.
Discussion: The results of this study link the historically mild phenotypes seen in individuals amenable to exon 8 skipping and with certain nonsense variations with alterations in trajectories of lower extremity muscle replacement by fat.
© 2022 American Academy of Neurology.
Figures
References
-
- Mendell JR, Moxley RT, Griggs RC, et al. Randomized, double-blind six-month trial of prednisone in Duchenne's muscular dystrophy. N Engl J Med. 1989,320(24):1592-1597. - PubMed
-
- McDonald CM, Henricson EK, Abresch RT, et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. The Lancet 2017,391(11019):451-461. - PubMed
-
- King WM, Ruttencutter R, Nagaraja HN, et al. Orthopedic outcomes of long-term daily corticosteroid treatment in Duchenne muscular dystrophy. Neurology 2007,68(19):1607-1613. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous