

Rare lysosomal disease registries: lessons learned over three decades of real-world evidence
- PMID: 36244992
- PMCID: PMC9573793
- DOI: 10.1186/s13023-022-02517-0
Rare lysosomal disease registries: lessons learned over three decades of real-world evidence
Abstract
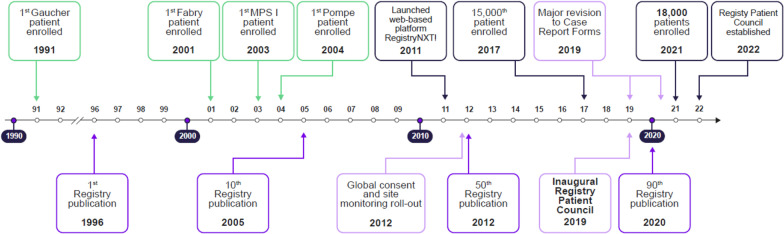
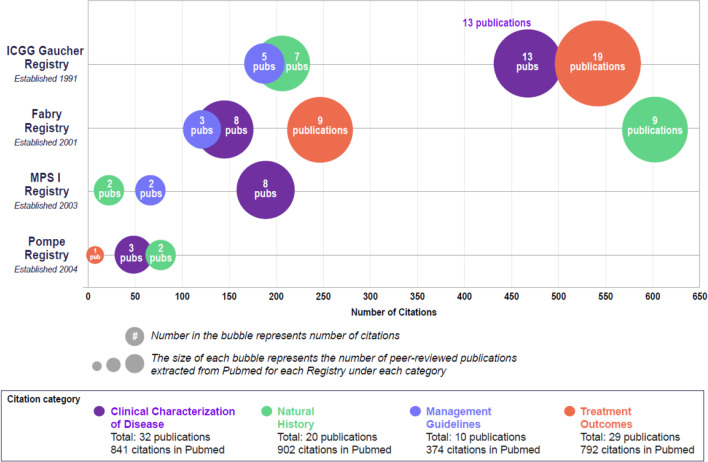
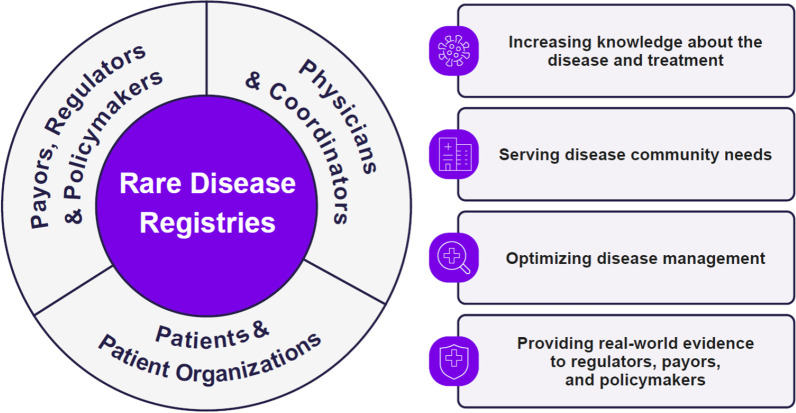
Lysosomal storage disorders (LSD) are rare diseases, caused by inherited deficiencies of lysosomal enzymes/transporters, that affect 1 in 7000 to 1 in 8000 newborns. Individuals with LSDs face long diagnostic journeys during which debilitating and life-threatening events can occur. Clinical trials and classical descriptions of LSDs typically focus on common manifestations, which are not representative of the vast phenotypic heterogeneity encountered in real-world experience. Additionally, recognizing that there was a limited understanding of the natural history, disease progression, and real-world clinical outcomes of rare LSDs, a collaborative partnership was pioneered 30 years ago to address these gaps. The Rare Disease Registries (RDR) (for Gaucher, Fabry, Mucopolysaccharidosis type I, and Pompe), represent the largest observational database for these LSDs. Over the past thirty years, data from the RDRs have helped to inform scientific understanding and the development of comprehensive monitoring and treatment guidelines by creating a framework for data collection and establishing a standard of care, with an overarching goal to improve the quality of life of affected patients. Here, we highlight the history, process, and impact of the RDRs, and discuss the lessons learned and future directions.
Keywords: Enzyme replacement therapy; Fabry; Gaucher; Lysosomal storage disorders; Mucopolysaccharidosis type I (MPS I); Pompe; Rare disease; Real-world data; Real-world evidence; Registries.
© 2022. The Author(s).
Conflict of interest statement
PKM has received research and travel support from Sanofi. PK has received research/grant support from Sanofi and Amicus Therapeutics, has received consulting fees and honoraria from Sanofi, Amicus Therapeutics, Maze Therapeutics, JCR Pharmaceutical and Asklepios Biopharmaceutical, Inc. (AskBio), is a member of the Pompe and Gaucher Disease Registry Advisory Board for Sanofi, Amicus Therapeutics, and Baebies, and has equity in Asklepios Biopharmaceutical, Inc. (AskBio), which is developing gene therapy for Pompe disease and Maze Therapeutics, which is developing a small molecule in Pompe disease. CW has received honoraria for board meetings and lecturing from Amicus, Chiesi, Idorsia, Sanofi, and Takeda. JB, JLB, DD, JF are employees of Sanofi and may hold Sanofi stocks.
Figures
References
-
- Fuller M, Meikle PJ, Hopwood JJ. Epidemiology of lysosomal storage diseases: an overview. In: Mehta A, Beck M, Sunder-Plassmann G (eds). Fabry Disease: Perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis Copyright © 2006, Oxford PharmaGenesis™. 2006.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
