

High detection rate for disease-causing variants in a cohort of 30 Iranian pediatric steroid resistant nephrotic syndrome cases
- PMID: 36245711
- PMCID: PMC9555279
- DOI: 10.3389/fped.2022.974840
High detection rate for disease-causing variants in a cohort of 30 Iranian pediatric steroid resistant nephrotic syndrome cases
Abstract
Background: Steroid resistant nephrotic syndrome (SRNS) represents a significant renal disease burden in childhood and adolescence. In contrast to steroid sensitive nephrotic syndrome (SSNS), renal outcomes are significantly poorer in SRNS. Over the past decade, extensive genetic heterogeneity has become evident while disease-causing variants are still only identified in 30% of cases in previously reported studies with proportion and type of variants identified differing depending on the age of onset and ethnical background of probands. A genetic diagnosis however can have implications regarding clinical management, including kidney transplantation, extrarenal disease manifestations, and, in some cases, even causal therapy. Genetic diagnostics therefore play an important role for the clinical care of SRNS affected individuals.
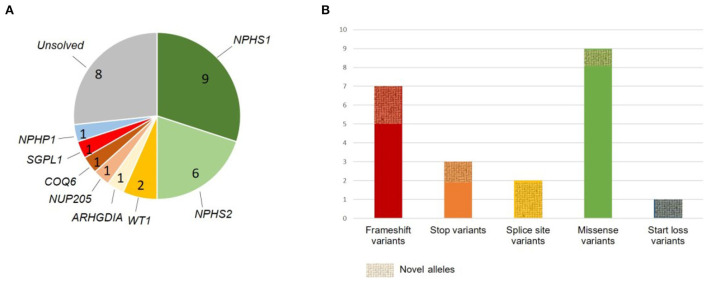
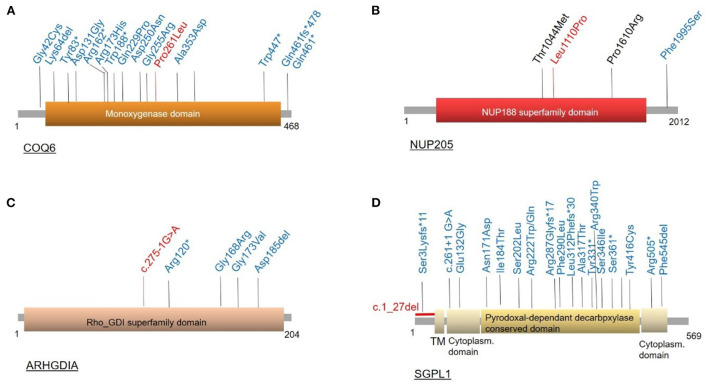
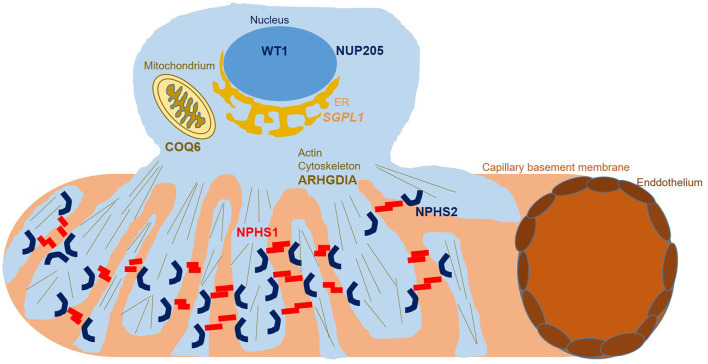
Methodology and results: Here, we performed NPHS2 Sanger sequencing and subsequent exome sequencing in 30 consanguineous Iranian families with a child affected by SRNS with a mean age of onset of 16 months. We identified disease-causing variants and one variant of uncertain significance in 22 families (73%), including variants in NPHS1 (30%), followed by NPHS2 (20%), WT1 (7%) as well as in NUP205, COQ6, ARHGDIA, SGPL1, and NPHP1 in single cases. Eight of these variants have not previously been reported as disease-causing, including four NPHS1 variants and one variant in NPHS2, ARHGDIA, SGPL1, and NPHP1 each.
Conclusion: In line with previous studies in non-Iranian subjects, we most frequently identified disease-causing variants in NPHS1 and NPHS2. While Sanger sequencing of NPHS2 can be considered as first diagnostic step in non-congenital cases, the genetic heterogeneity underlying SRNS renders next-generation sequencing based diagnostics as the most efficient genetic screening method. In accordance with the mainly autosomal recessive inheritance pattern, diagnostic yield can be significantly higher in consanguineous than in outbred populations.
Keywords: ARHGDIA; COQ6; Iran; NUP205; SGPL1; SRNS; nephrotic syndrome.
Copyright © 2022 Najafi, Riedhammer, Rad, Torbati, Berutti, Schüle, Schroda, Meitinger, Ćomić, Bojd, Baranzehi, Shojaei, Azarfar, Khazaei, Köttgen, Backofen, Karimiani, Hoefele and Schmidts.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Genetic analysis and outcomes of Omani children with steroid-resistant nephrotic syndrome.Mol Genet Genomic Med. 2023 Sep;11(9):e2201. doi: 10.1002/mgg3.2201. Epub 2023 May 19. Mol Genet Genomic Med. 2023. PMID: 37204080 Free PMC article.
-
NPHS2 mutation analysis shows genetic heterogeneity of steroid-resistant nephrotic syndrome and low post-transplant recurrence.Kidney Int. 2004 Aug;66(2):571-9. doi: 10.1111/j.1523-1755.2004.00776.x. Kidney Int. 2004. PMID: 15253708
-
Novel NPHS2 variant in patients with familial steroid-resistant nephrotic syndrome with early onset, slow progression and dominant inheritance pattern.Clin Exp Nephrol. 2017 Aug;21(4):677-684. doi: 10.1007/s10157-016-1331-3. Epub 2016 Aug 29. Clin Exp Nephrol. 2017. PMID: 27573339
-
Steroid Resistant Nephrotic Syndrome-Genetic Consideration.Pril (Makedon Akad Nauk Umet Odd Med Nauki). 2015;36(3):5-12. doi: 10.1515/prilozi-2015-0073. Pril (Makedon Akad Nauk Umet Odd Med Nauki). 2015. PMID: 27442391 Review.
-
Exploring the Clinical and Genetic Spectrum of Steroid Resistant Nephrotic Syndrome: The PodoNet Registry.Front Pediatr. 2018 Jul 17;6:200. doi: 10.3389/fped.2018.00200. eCollection 2018. Front Pediatr. 2018. PMID: 30065916 Free PMC article. Review.
Cited by
-
Nucleoporin-associated steroid-resistant nephrotic syndrome.Pediatr Nephrol. 2025 Mar;40(3):629-649. doi: 10.1007/s00467-024-06494-3. Epub 2024 Sep 27. Pediatr Nephrol. 2025. PMID: 39331077 Review.
-
Case Report: a novel variant in WT1 leads to focal segmental glomerulosclerosis and uterovaginal anomalies through exon skipping.Front Nephrol. 2025 Apr 1;5:1542475. doi: 10.3389/fneph.2025.1542475. eCollection 2025. Front Nephrol. 2025. PMID: 40235736 Free PMC article.
-
Factors influencing survival in sphingosine phosphate lyase insufficiency syndrome: a retrospective cross-sectional natural history study of 76 patients.Orphanet J Rare Dis. 2024 Sep 27;19(1):355. doi: 10.1186/s13023-024-03311-w. Orphanet J Rare Dis. 2024. PMID: 39334450 Free PMC article.
-
NPHS Mutations in Pediatric Patients with Congenital and Steroid-Resistant Nephrotic Syndrome.Int J Mol Sci. 2024 Nov 15;25(22):12275. doi: 10.3390/ijms252212275. Int J Mol Sci. 2024. PMID: 39596340 Free PMC article. Review.
-
Prevalence estimate of sphingosine phosphate lyase insufficiency syndrome in worldwide and select populations.Genet Med Open. 2023 Oct 30;2:100840. doi: 10.1016/j.gimo.2023.100840. eCollection 2024. Genet Med Open. 2023. PMID: 39669624 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Miscellaneous