

Prognostic Modeling of Lung Adenocarcinoma Based on Hypoxia and Ferroptosis-Related Genes
- PMID: 36245988
- PMCID: PMC9553523
- DOI: 10.1155/2022/1022580
Prognostic Modeling of Lung Adenocarcinoma Based on Hypoxia and Ferroptosis-Related Genes
Abstract
Background: It is well known that hypoxia and ferroptosis are intimately connected with tumor development. The purpose of this investigation was to identify whether they have a prognostic signature. To this end, genes related to hypoxia and ferroptosis scores were investigated using bioinformatics analysis to stratify the risk of lung adenocarcinoma.
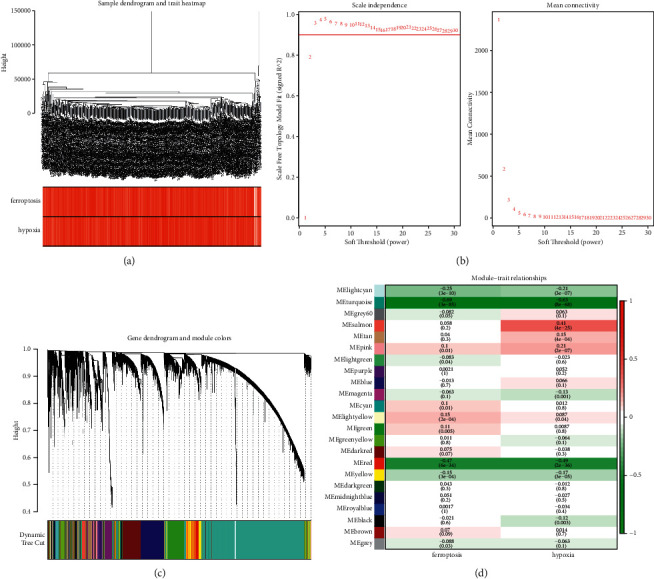
Methods: Hypoxia and ferroptosis scores were estimated using The Cancer Genome Atlas (TCGA) database-derived cohort transcriptome profiles via the single sample gene set enrichment analysis (ssGSEA) algorithm. The candidate genes associated with hypoxia and ferroptosis scores were identified using weighted correlation network analysis (WGCNA) and differential expression analysis. The prognostic genes in this study were discovered using the Cox regression (CR) model in conjunction with the LASSO method, which was then utilized to create a prognostic signature. The efficacy, accuracy, and clinical value of the prognostic model were evaluated using an independent validation cohort, Receiver Operator Characteristic (ROC) curve, and nomogram. The analysis of function and immune cell infiltration was also carried out.
Results: Here, we appraised 152 candidate genes expressed not the same, which were related to hypoxia and ferroptosis for prognostic modeling in The Cancer Genome Atlas Lung Adenocarcinoma (TCGA-LUAD) cohort, and these genes were further validated in the GSE31210 cohort. We found that the 14-gene-based prognostic model, utilizing MAPK4, TNS4, WFDC2, FSTL3, ITGA2, KLK11, PHLDB2, VGLL3, SNX30, KCNQ3, SMAD9, ANGPTL4, LAMA3, and STK32A, performed well in predicting the prognosis in lung adenocarcinoma. ROC and nomogram analyses showed that risk scores based on prognostic signatures provided desirable predictive accuracy and clinical utility. Moreover, gene set variance analysis showed differential enrichment of 33 hallmark gene sets between different risk groups. Additionally, our results indicated that a higher risk score will lead to more fibroblasts and activated CD4 T cells but fewer myeloid dendritic cells, endothelial cells, eosinophils, immature dendritic cells, and neutrophils.
Conclusion: Our research found a 14-gene signature and established a nomogram that accurately predicted the prognosis in patients with lung adenocarcinoma. Clinical decision-making and therapeutic customization may benefit from these results, which may serve as a valuable reference in the future.
Copyright © 2022 Chang Liu et al.
Conflict of interest statement
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Figures










References
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
