

Integration of mRNA and microRNA analysis reveals the molecular mechanisms underlying drought stress tolerance in maize (Zea mays L.)
- PMID: 36247625
- PMCID: PMC9557922
- DOI: 10.3389/fpls.2022.932667
Integration of mRNA and microRNA analysis reveals the molecular mechanisms underlying drought stress tolerance in maize (Zea mays L.)
Abstract
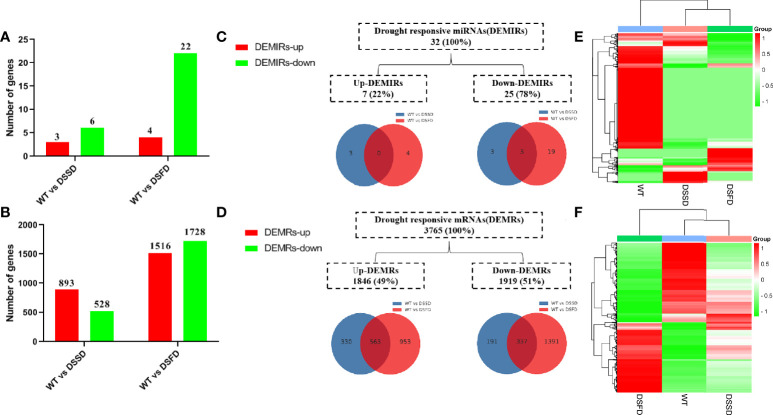
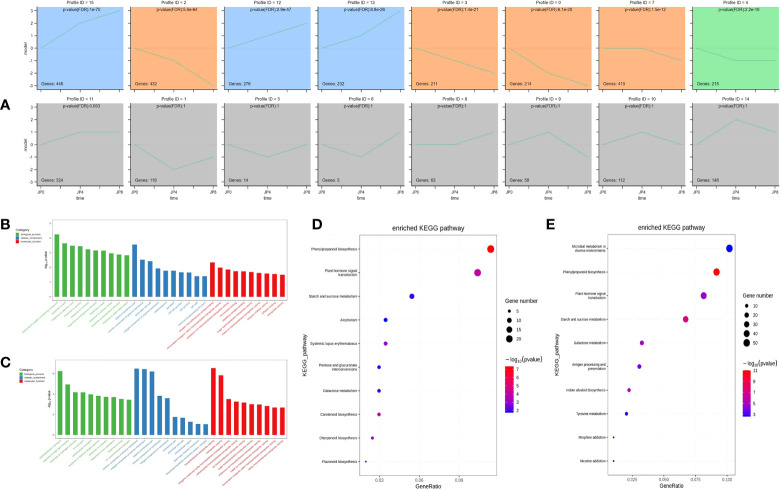
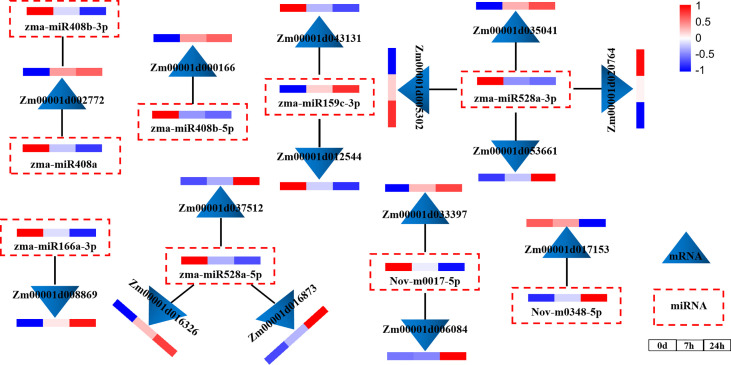
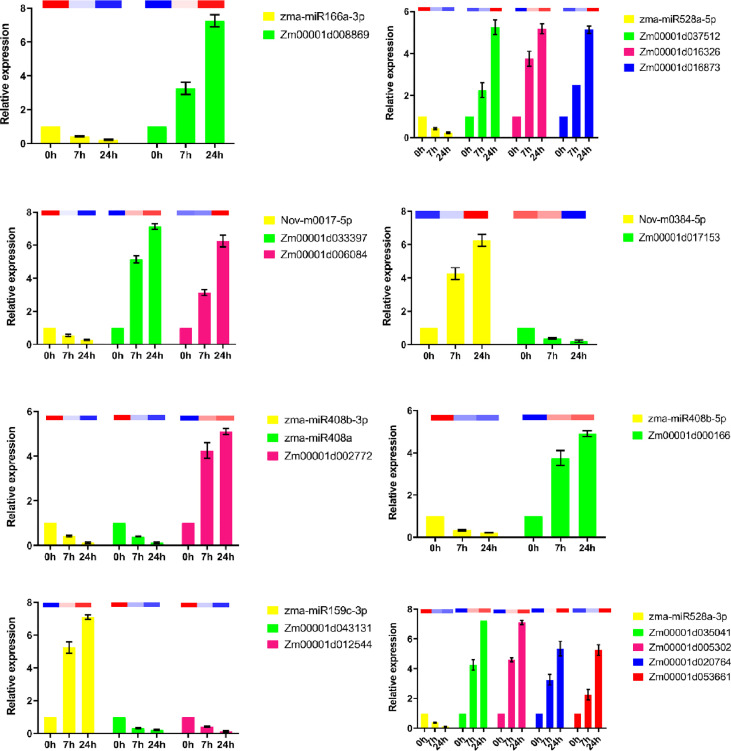
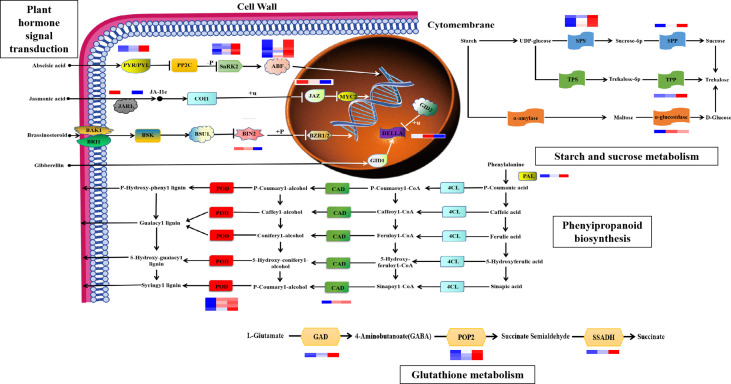
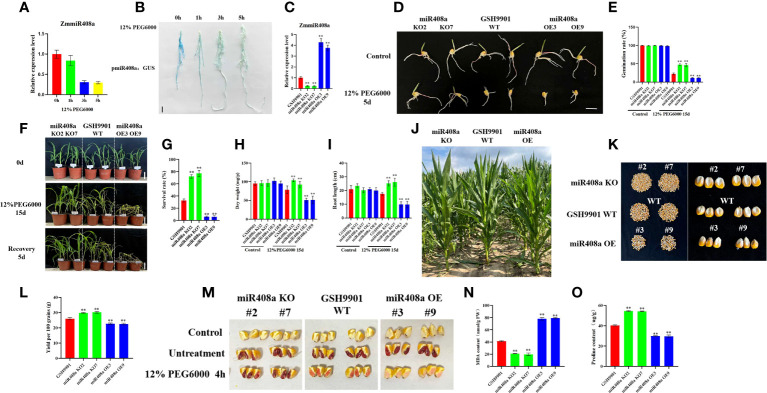
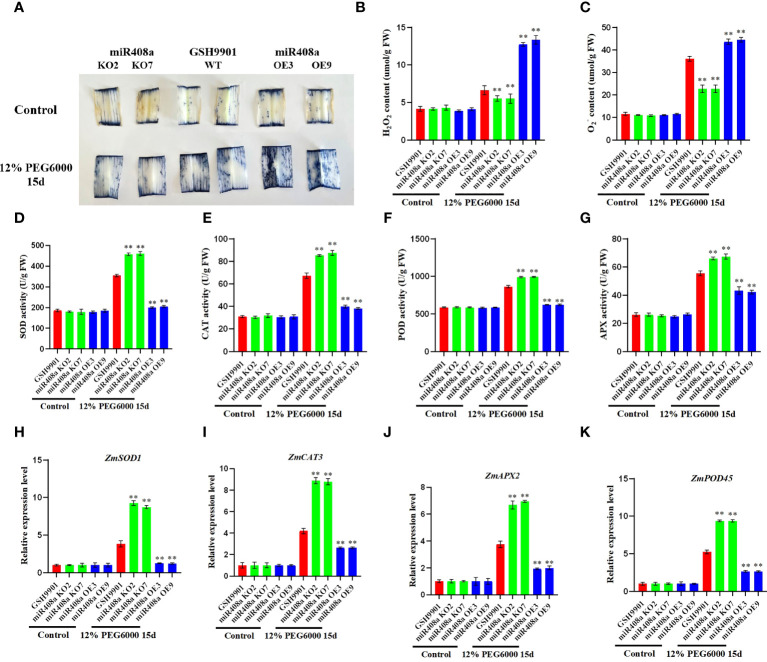
Drought is among the most serious environmental issue globally, and seriously affects the development, growth, and yield of crops. Maize (Zea mays L.), an important crop and industrial raw material, is planted on a large scale worldwide and drought can lead to large-scale reductions in maize corn production; however, few studies have focused on the maize root system mechanisms underlying drought resistance. In this study, miRNA-mRNA analysis was performed to deeply analyze the molecular mechanisms involved in drought response in the maize root system under drought stress. Furthermore, preliminary investigation of the biological function of miR408a in the maize root system was also conducted. The morphological, physiological, and transcriptomic changes in the maize variety "M8186" at the seedling stage under 12% PEG 6000 drought treatment (0, 7, and 24 h) were analyzed. With prolonged drought stress, seedlings gradually withered, the root system grew significantly, and abscisic acid, brassinolide, lignin, glutathione, and trehalose content in the root system gradually increased. Furthermore, peroxidase activity increased, while gibberellic acid and jasmonic acid gradually decreased. Moreover, 32 differentially expressed miRNAs (DEMIRs), namely, 25 known miRNAs and 7 new miRNAs, and 3,765 differentially expressed mRNAs (DEMRs), were identified in maize root under drought stress by miRNA-seq and mRNA-seq analysis, respectively. Through combined miRNA-mRNA analysis, 16 miRNA-target gene pairs, comprising 9 DEMIRs and 15 DEMRs, were obtained. In addition, four metabolic pathways, namely, "plant hormone signal transduction", "phenylpropane biosynthesis", "glutathione metabolism", and "starch and sucrose metabolism", were predicted to have important roles in the response of the maize root system to drought. MiRNA and mRNA expression results were verified by real-time quantitative PCR. Finally, miR408a was selected for functional analysis and demonstrated to be a negative regulator of drought response, mainly through regulation of reactive oxygen species accumulation in the maize root system. This study helps to elaborate the regulatory response mechanisms of the maize root system under drought stress and predicts the biological functions of candidate miRNAs and mRNAs, providing strategies for subsequent mining for, and biological breeding to select for, drought-responsive genes in the maize root system.
Keywords: drought stress; mRNA; maize; miR408a; microRNA.
Copyright © 2022 Jiao, Ma, Wang, Chen, Liu, Qu, Guan and Ma.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

Similar articles
-
Integrated analysis of transcriptome, sRNAome, and degradome involved in the drought-response of maize Zhengdan958.Open Life Sci. 2025 Jan 27;20(1):20221044. doi: 10.1515/biol-2022-1044. eCollection 2025. Open Life Sci. 2025. PMID: 39881824 Free PMC article.
-
Integration of mRNA and miRNA analysis reveals the molecular mechanism of potato (Solanum tuberosum L.) response to alkali stress.Int J Biol Macromol. 2021 Jul 1;182:938-949. doi: 10.1016/j.ijbiomac.2021.04.094. Epub 2021 Apr 18. Int J Biol Macromol. 2021. PMID: 33878362
-
Comparative transcriptomic analysis of contrasting hybrid cultivars reveal key drought-responsive genes and metabolic pathways regulating drought stress tolerance in maize at various stages.PLoS One. 2020 Oct 15;15(10):e0240468. doi: 10.1371/journal.pone.0240468. eCollection 2020. PLoS One. 2020. PMID: 33057352 Free PMC article.
-
Transcriptional regulatory networks in response to drought stress and rewatering in maize (Zea mays L.).Mol Genet Genomics. 2021 Nov;296(6):1203-1219. doi: 10.1007/s00438-021-01820-y. Epub 2021 Oct 3. Mol Genet Genomics. 2021. PMID: 34601650 Review.
-
Transcriptome expression profiles reveal response mechanisms to drought and drought-stress mitigation mechanisms by exogenous glycine betaine in maize.Biotechnol Lett. 2022 Mar;44(3):367-386. doi: 10.1007/s10529-022-03221-6. Epub 2022 Mar 16. Biotechnol Lett. 2022. PMID: 35294695 Review.
Cited by
-
Identification and analysis of differentially expressed trihelix genes in maize (Zea mays) under abiotic stresses.PeerJ. 2023 May 1;11:e15312. doi: 10.7717/peerj.15312. eCollection 2023. PeerJ. 2023. PMID: 37151290 Free PMC article.
-
Genomic variation, environmental adaptation, and feralization in ramie, an ancient fiber crop.Plant Commun. 2024 Aug 12;5(8):100942. doi: 10.1016/j.xplc.2024.100942. Epub 2024 May 8. Plant Commun. 2024. PMID: 38720463 Free PMC article.
-
A Novel Senescence-Specific Gene (ZmSAG39) Negatively Regulates Darkness and Drought Responses in Maize.Int J Mol Sci. 2022 Dec 15;23(24):15984. doi: 10.3390/ijms232415984. Int J Mol Sci. 2022. PMID: 36555622 Free PMC article.
-
Global knowledge mapping and emerging research trends in non-coding RNAs related to animal and plant male sterility: A visual analysis of CiteSpace maps.Medicine (Baltimore). 2025 Jun 6;104(23):e42612. doi: 10.1097/MD.0000000000042612. Medicine (Baltimore). 2025. PMID: 40489841 Free PMC article. Review.
-
The miRNA-mRNA Regulatory Modules of Pinus massoniana Lamb. in Response to Drought Stress.Int J Mol Sci. 2023 Sep 28;24(19):14655. doi: 10.3390/ijms241914655. Int J Mol Sci. 2023. PMID: 37834103 Free PMC article.
References
LinkOut - more resources
Full Text Sources