

Creating MHC-Restricted Neoantigens with Covalent Inhibitors That Can Be Targeted by Immune Therapy
- PMID: 36250888
- PMCID: PMC9827112
- DOI: 10.1158/2159-8290.CD-22-1074
Creating MHC-Restricted Neoantigens with Covalent Inhibitors That Can Be Targeted by Immune Therapy
Erratum in
-
Correction: Creating MHC-Restricted Neoantigens with Covalent Inhibitors That Can Be Targeted by Immune Therapy.Cancer Discov. 2023 Apr 3;13(4):1026. doi: 10.1158/2159-8290.CD-23-0125. Cancer Discov. 2023. PMID: 37009704 Free PMC article. No abstract available.
Abstract
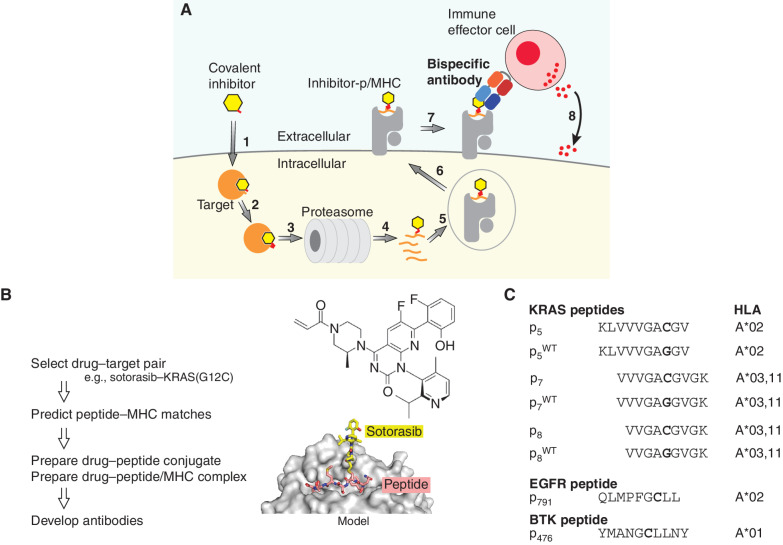
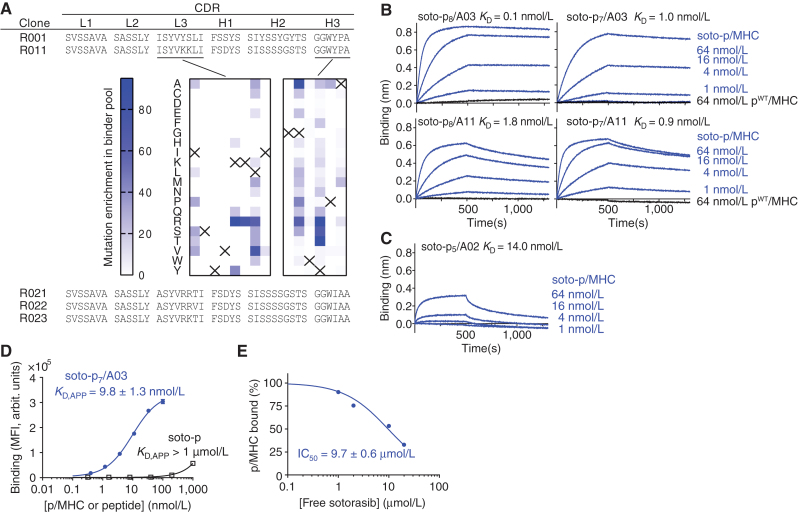
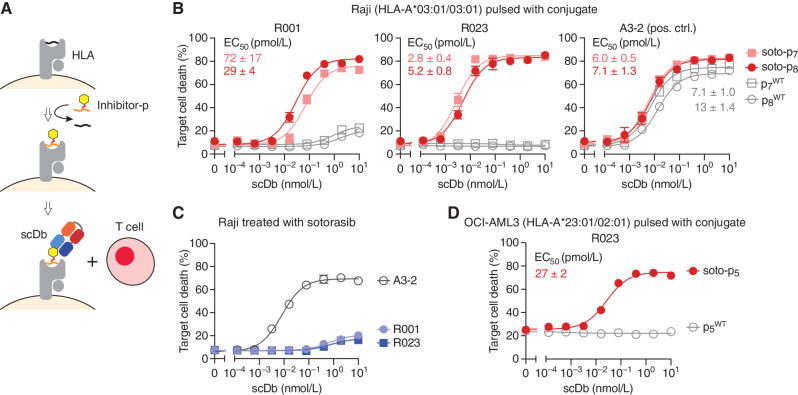
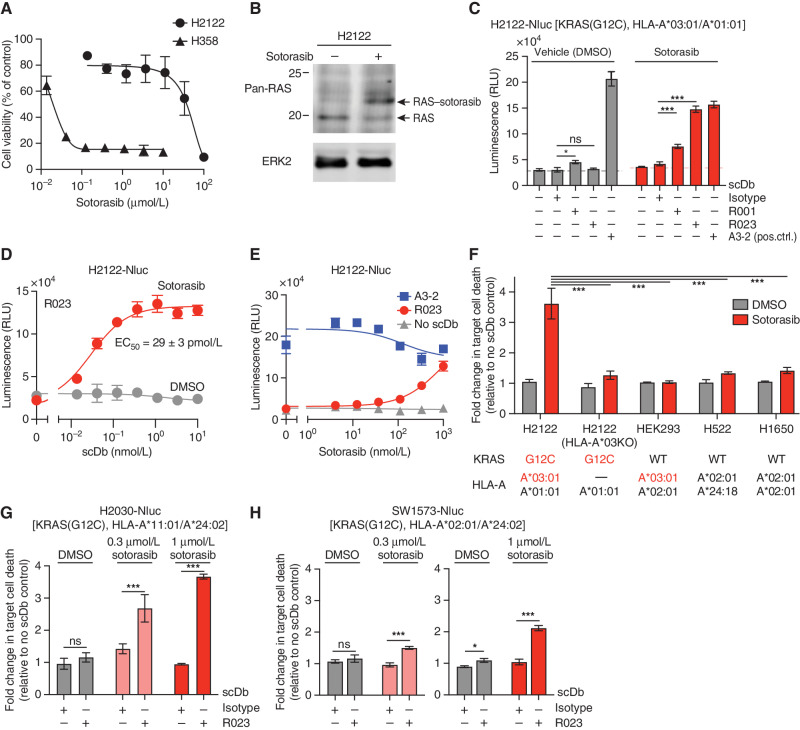
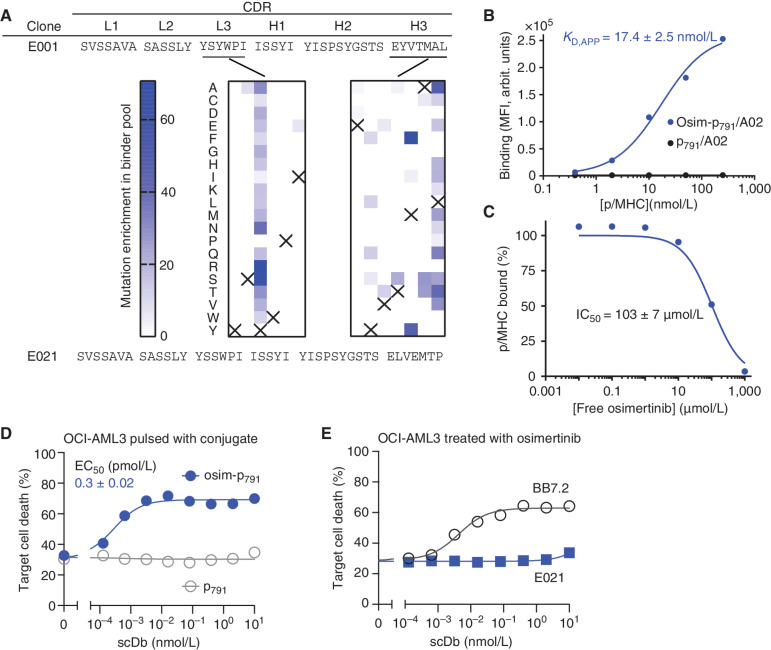
Intracellular oncoproteins can be inhibited with targeted therapy, but responses are not durable. Immune therapies can be curative, but most oncogene-driven tumors are unresponsive to these agents. Fragments of intracellular oncoproteins can act as neoantigens presented by the major histocompatibility complex (MHC), but recognizing minimal differences between oncoproteins and their normal counterparts is challenging. We have established a platform technology that exploits hapten-peptide conjugates generated by covalent inhibitors to create distinct neoantigens that selectively mark cancer cells. Using the FDA-approved covalent inhibitors sotorasib and osimertinib, we developed "HapImmune" antibodies that bind to drug-peptide conjugate/MHC complexes but not to the free drugs. A HapImmune-based bispecific T-cell engager selectively and potently kills sotorasib-resistant lung cancer cells upon sotorasib treatment. Notably, it is effective against KRASG12C-mutant cells with different HLA supertypes, HLA-A*02 and A*03/11, suggesting loosening of MHC restriction. Our strategy creates targetable neoantigens by design, unifying targeted and immune therapies.
Significance: Targeted therapies against oncoproteins often have dramatic initial efficacy but lack durability. Immunotherapies can be curative, yet most tumors fail to respond. We developed a generalizable technology platform that exploits hapten-peptides generated by covalent inhibitors as neoantigens presented on MHC to enable engineered antibodies to selectively kill drug-resistant cancer cells. See related commentary by Cox et al., p. 19. This article is highlighted in the In This Issue feature, p. 1.
©2022 The Authors; Published by the American Association for Cancer Research.
Figures
Comment in
-
Bispecific T cell engagers kill resistant cells during KRAS-G12C blockade therapy.Oncoimmunology. 2022 Nov 1;11(1):2141978. doi: 10.1080/2162402X.2022.2141978. eCollection 2022. Oncoimmunology. 2022. PMID: 36338145 Free PMC article.
-
Convergence of Targeted and Immune Therapies for the Treatment of Oncogene-Driven Cancers.Cancer Discov. 2023 Jan 9;13(1):19-22. doi: 10.1158/2159-8290.CD-22-1199. Cancer Discov. 2023. PMID: 36620884
References
-
- Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. . The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019;575:217–23. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
