

The 5-Hydroxymethylcytosine Landscape of Prostate Cancer
- PMID: 36251389
- PMCID: PMC9627125
- DOI: 10.1158/0008-5472.CAN-22-1123
The 5-Hydroxymethylcytosine Landscape of Prostate Cancer
Abstract
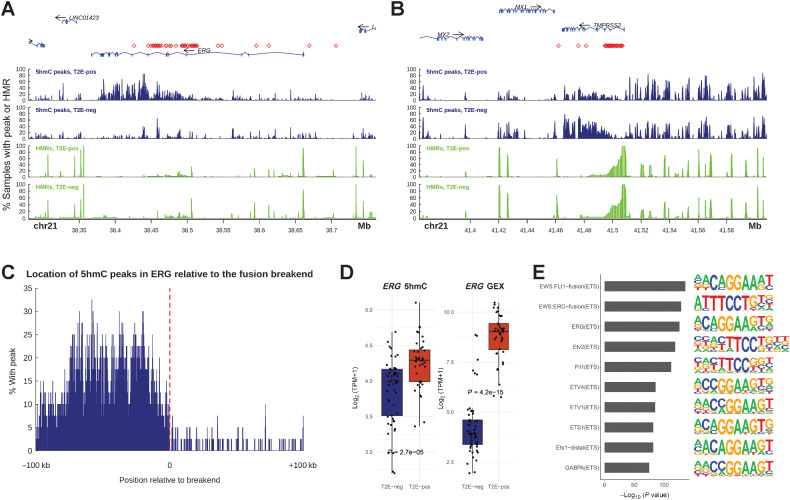
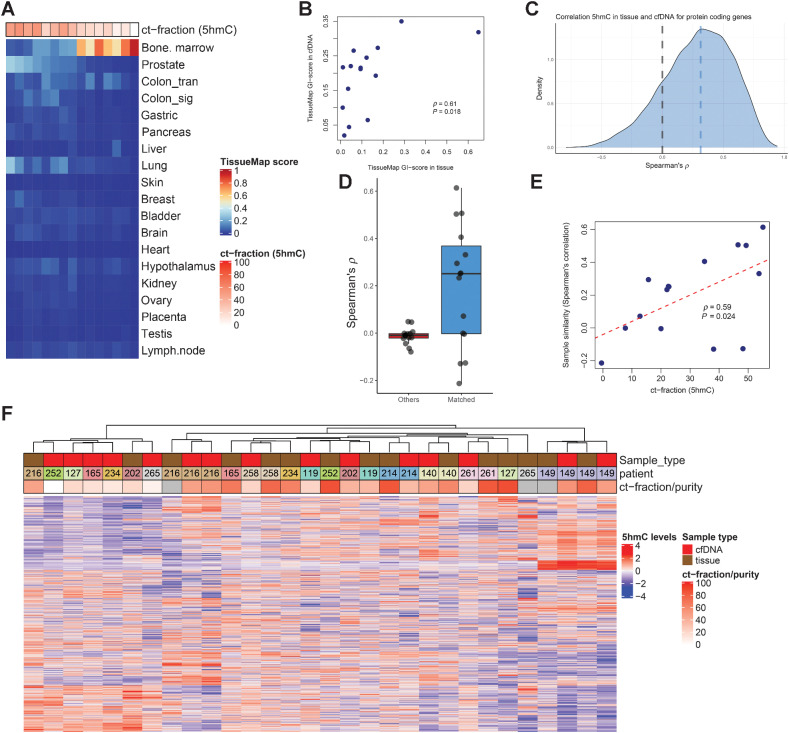
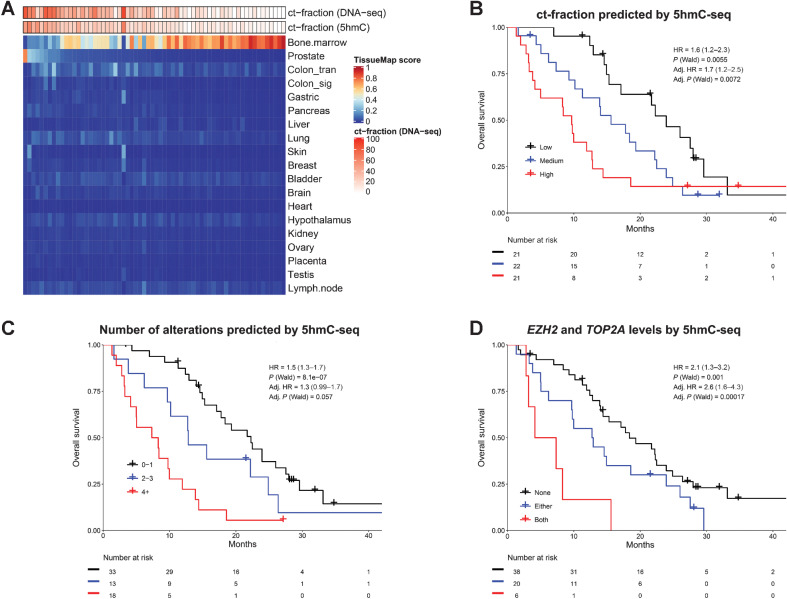
Analysis of DNA methylation is a valuable tool to understand disease progression and is increasingly being used to create diagnostic and prognostic clinical biomarkers. While conversion of cytosine to 5-methylcytosine (5mC) commonly results in transcriptional repression, further conversion to 5-hydroxymethylcytosine (5hmC) is associated with transcriptional activation. Here we perform the first study integrating whole-genome 5hmC with DNA, 5mC, and transcriptome sequencing in clinical samples of benign, localized, and advanced prostate cancer. 5hmC is shown to mark activation of cancer drivers and downstream targets. Furthermore, 5hmC sequencing revealed profoundly altered cell states throughout the disease course, characterized by increased proliferation, oncogenic signaling, dedifferentiation, and lineage plasticity to neuroendocrine and gastrointestinal lineages. Finally, 5hmC sequencing of cell-free DNA from patients with metastatic disease proved useful as a prognostic biomarker able to identify an aggressive subtype of prostate cancer using the genes TOP2A and EZH2, previously only detectable by transcriptomic analysis of solid tumor biopsies. Overall, these findings reveal that 5hmC marks epigenomic activation in prostate cancer and identify hallmarks of prostate cancer progression with potential as biomarkers of aggressive disease.
Significance: In prostate cancer, 5-hydroxymethylcytosine delineates oncogene activation and stage-specific cell states and can be analyzed in liquid biopsies to detect cancer phenotypes. See related article by Wu and Attard, p. 3880.
©2022 The Authors; Published by the American Association for Cancer Research.
Figures
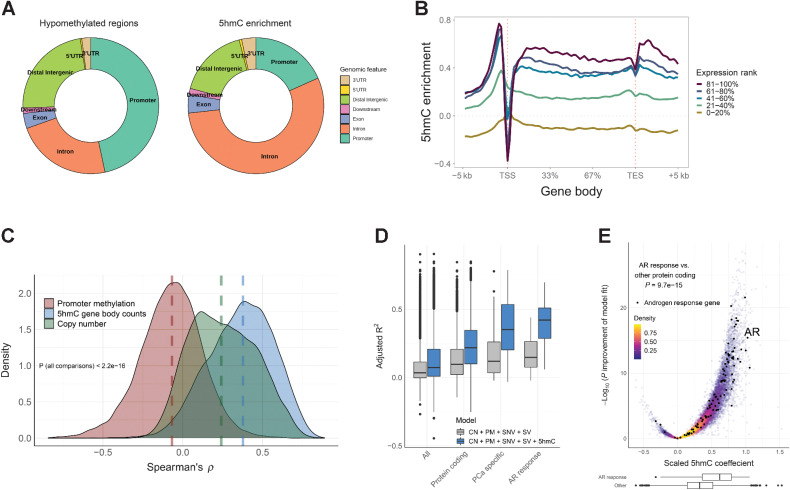
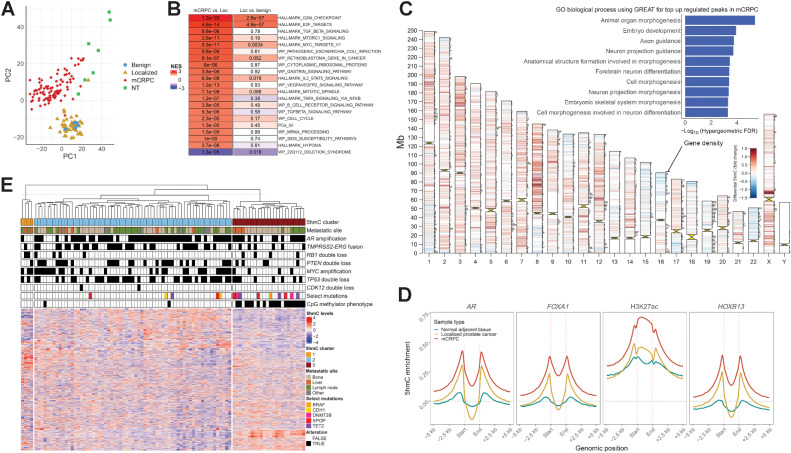
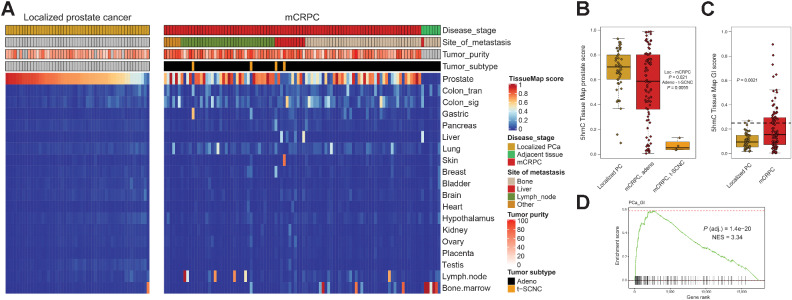
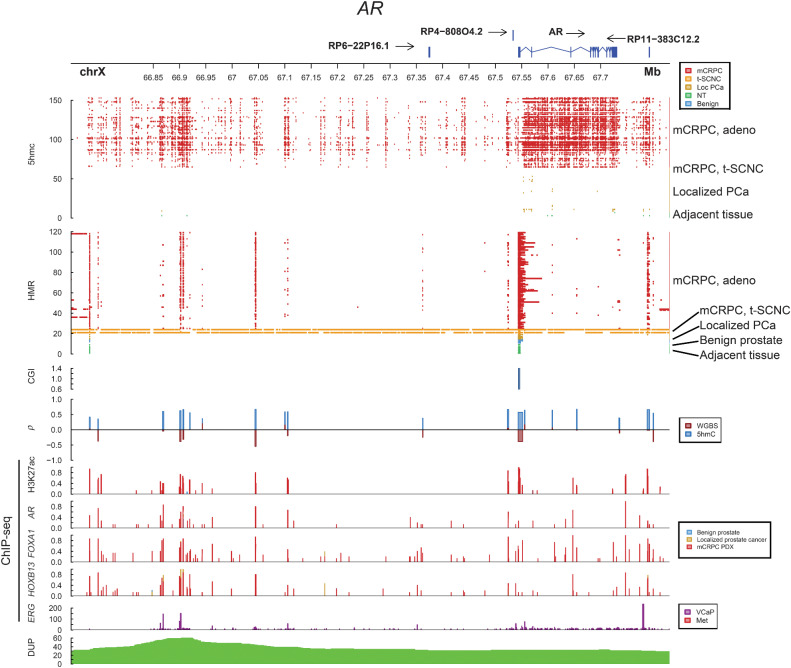
Comment in
-
Accidentals of the DNA Symphony.Cancer Res. 2022 Nov 2;82(21):3880-3881. doi: 10.1158/0008-5472.CAN-22-2750. Cancer Res. 2022. PMID: 36321266
References
Publication types
MeSH terms
Substances
Grants and funding
- P30 CA093373/CA/NCI NIH HHS/United States
- R01 CA227025/CA/NCI NIH HHS/United States
- R01 CA251245/CA/NCI NIH HHS/United States
- P50 CA097186/CA/NCI NIH HHS/United States
- R01 CA208258/CA/NCI NIH HHS/United States
- R01 CA230516/CA/NCI NIH HHS/United States
- R01 HG012227/HG/NHGRI NIH HHS/United States
- P30 CA016042/CA/NCI NIH HHS/United States
- K08 CA226348/CA/NCI NIH HHS/United States
- P50 CA186786/CA/NCI NIH HHS/United States
- R01 HG006827/HG/NHGRI NIH HHS/United States
- U01 CA214194/CA/NCI NIH HHS/United States
- DP2 CA239597/CA/NCI NIH HHS/United States
- DP2 CA271832/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
