

Computational Approaches and Challenges in Spatial Transcriptomics
- PMID: 36252814
- PMCID: PMC10372921
- DOI: 10.1016/j.gpb.2022.10.001
Computational Approaches and Challenges in Spatial Transcriptomics
Abstract
The development of spatial transcriptomics (ST) technologies has transformed genetic research from a single-cell data level to a two-dimensional spatial coordinate system and facilitated the study of the composition and function of various cell subsets in different environments and organs. The large-scale data generated by these ST technologies, which contain spatial gene expression information, have elicited the need for spatially resolved approaches to meet the requirements of computational and biological data interpretation. These requirements include dealing with the explosive growth of data to determine the cell-level and gene-level expression, correcting the inner batch effect and loss of expression to improve the data quality, conducting efficient interpretation and in-depth knowledge mining both at the single-cell and tissue-wide levels, and conducting multi-omics integration analysis to provide an extensible framework toward the in-depth understanding of biological processes. However, algorithms designed specifically for ST technologies to meet these requirements are still in their infancy. Here, we review computational approaches to these problems in light of corresponding issues and challenges, and present forward-looking insights into algorithm development.
Keywords: Computational approach; Data interpretation; Data quality; Multi-omics integration; Spatial transcriptomics.
Copyright © 2023 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
All authors are current employees of BGI Group Ltd.
Figures

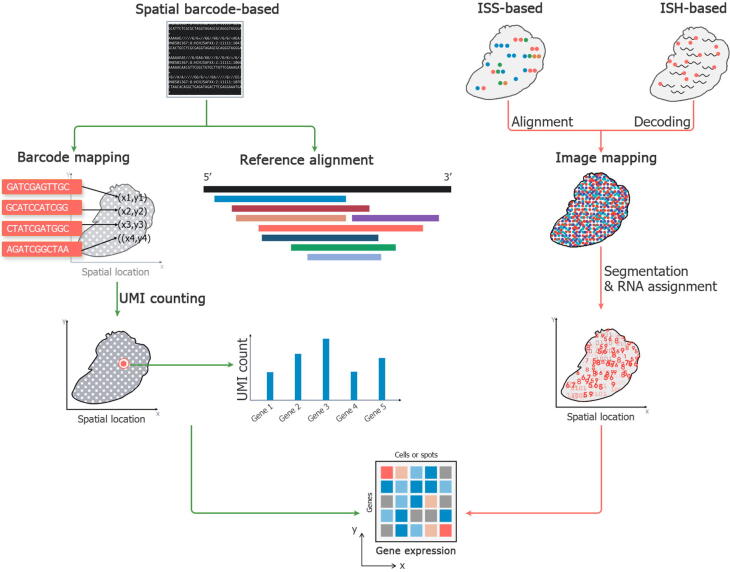

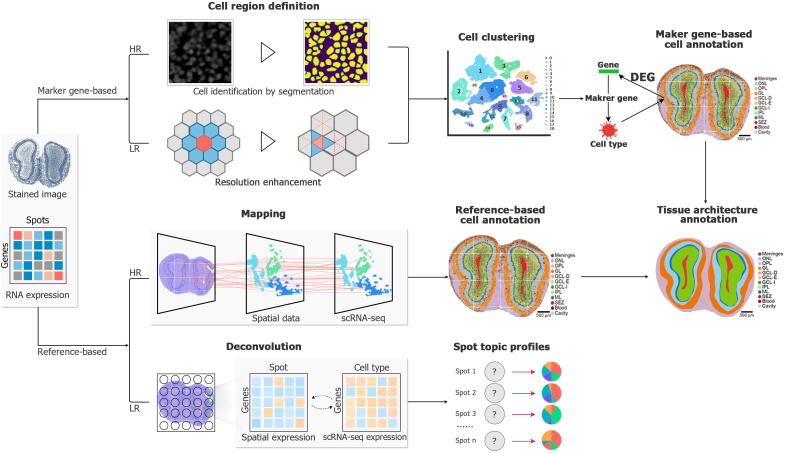

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
