

The synthetic lethality of targeting cell cycle checkpoints and PARPs in cancer treatment
- PMID: 36253861
- PMCID: PMC9578258
- DOI: 10.1186/s13045-022-01360-x
The synthetic lethality of targeting cell cycle checkpoints and PARPs in cancer treatment
Abstract
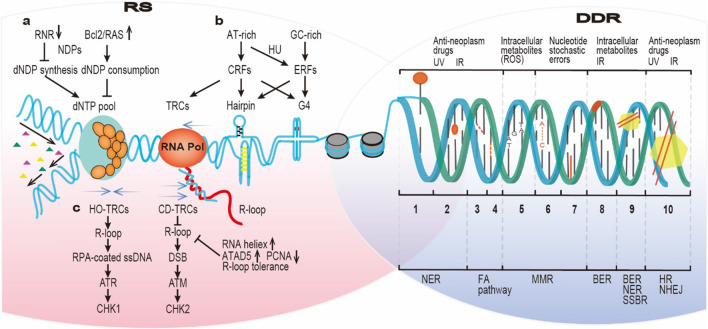
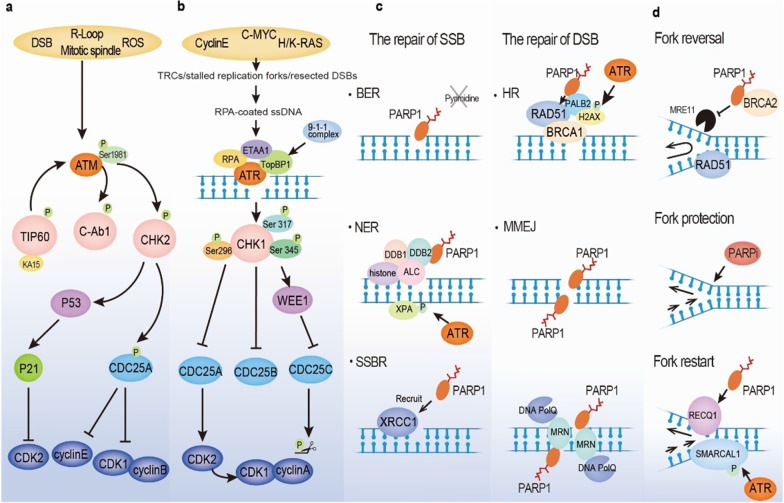
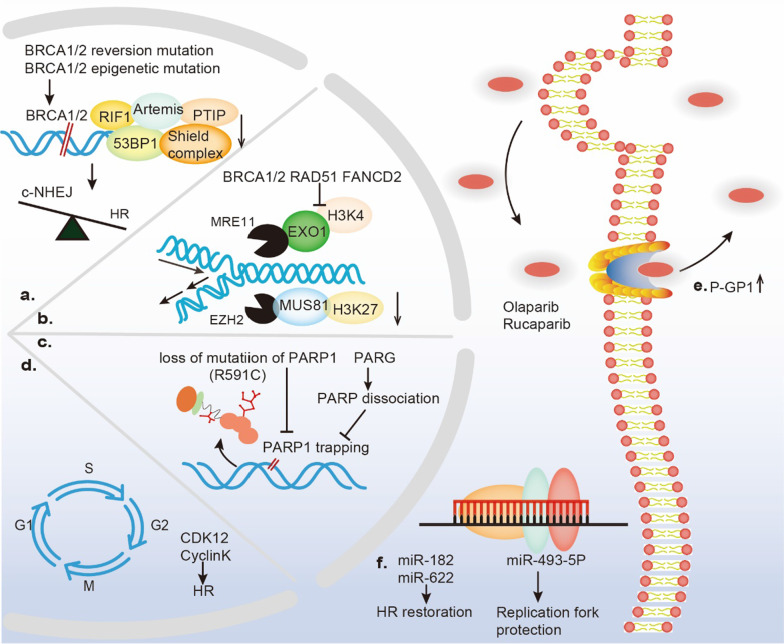
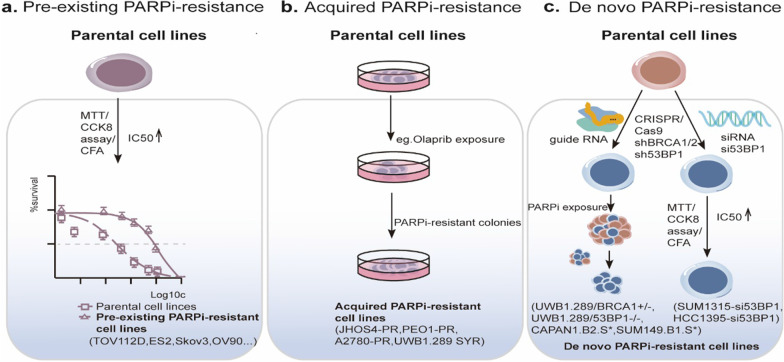
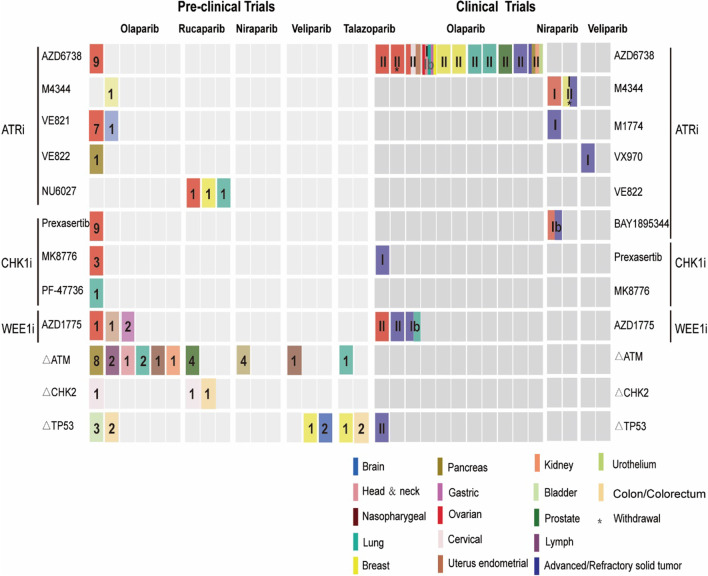
Continuous cell division is a hallmark of cancer, and the underlying mechanism is tumor genomics instability. Cell cycle checkpoints are critical for enabling an orderly cell cycle and maintaining genome stability during cell division. Based on their distinct functions in cell cycle control, cell cycle checkpoints are classified into two groups: DNA damage checkpoints and DNA replication stress checkpoints. The DNA damage checkpoints (ATM-CHK2-p53) primarily monitor genetic errors and arrest cell cycle progression to facilitate DNA repair. Unfortunately, genes involved in DNA damage checkpoints are frequently mutated in human malignancies. In contrast, genes associated with DNA replication stress checkpoints (ATR-CHK1-WEE1) are rarely mutated in tumors, and cancer cells are highly dependent on these genes to prevent replication catastrophe and secure genome integrity. At present, poly (ADP-ribose) polymerase inhibitors (PARPi) operate through "synthetic lethality" mechanism with mutant DNA repair pathways genes in cancer cells. However, an increasing number of patients are acquiring PARP inhibitor resistance after prolonged treatment. Recent work suggests that a combination therapy of targeting cell cycle checkpoints and PARPs act synergistically to increase the number of DNA errors, compromise the DNA repair machinery, and disrupt the cell cycle, thereby increasing the death rate of cancer cells with DNA repair deficiency or PARP inhibitor resistance. We highlight a combinational strategy involving PARP inhibitors and inhibition of two major cell cycle checkpoint pathways, ATM-CHK2-TP53 and ATR-CHK1-WEE1. The biological functions, resistance mechanisms against PARP inhibitors, advances in preclinical research, and clinical trials are also reviewed.
Keywords: Cancer; Cell cycle checkpoint; Drug resistance; PARP inhibitors; Synthetic lethality; Targeted therapy.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Kotsantis P, Petermann E, Boulton SJ. Mechanisms of oncogene-induced replication stress: jigsaw falling into place. Cancer Discov. 2018;8(5):537–555. doi: 10.1158/2159-8290.CD-17-1461. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
