

Host genetic factors related to innate immunity, environmental sensing and cellular functions are associated with human skin microbiota
- PMID: 36261456
- PMCID: PMC9582029
- DOI: 10.1038/s41467-022-33906-5
Host genetic factors related to innate immunity, environmental sensing and cellular functions are associated with human skin microbiota
Abstract
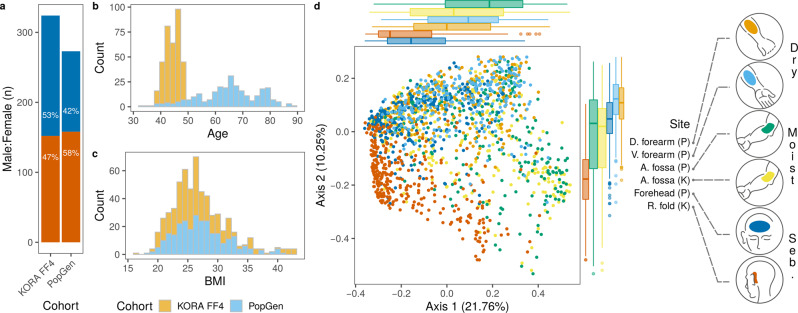
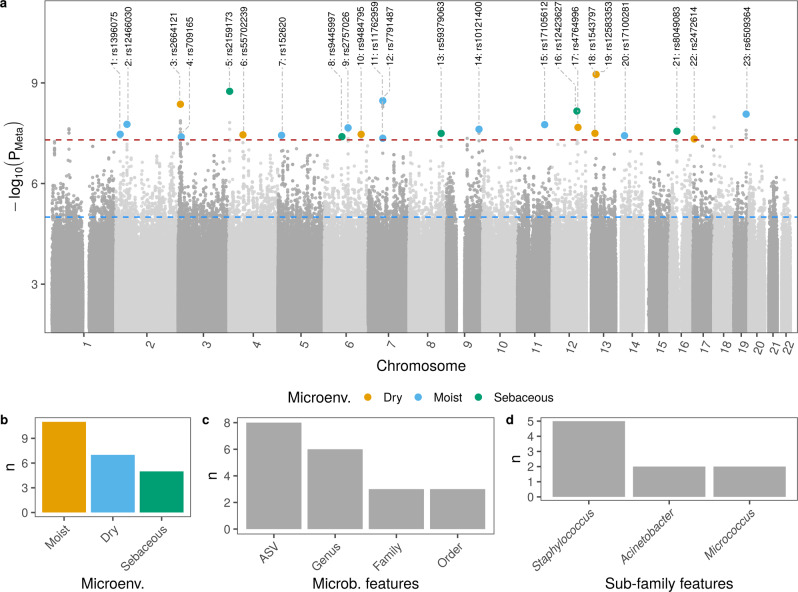
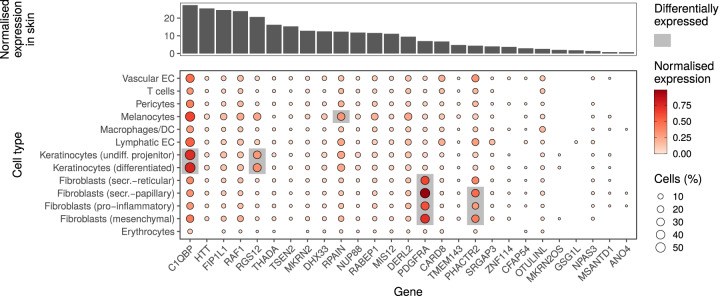
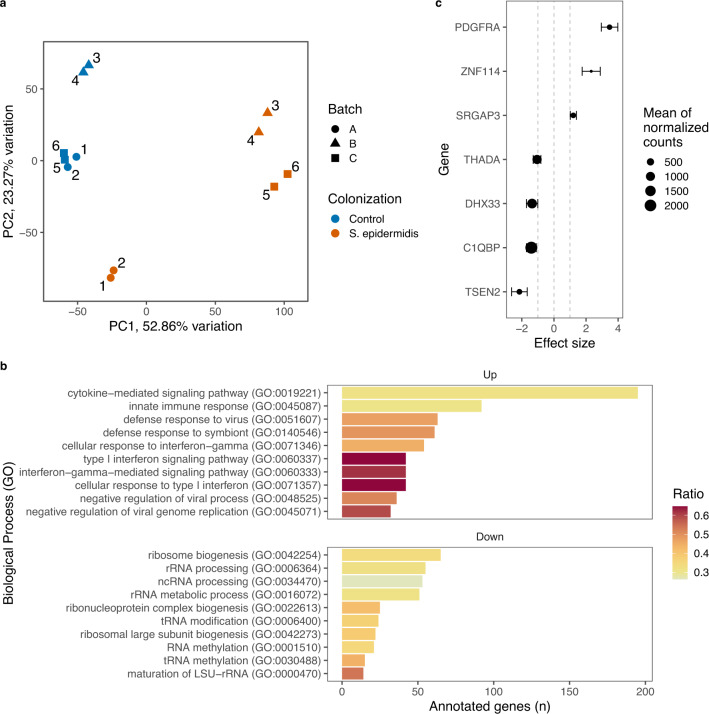
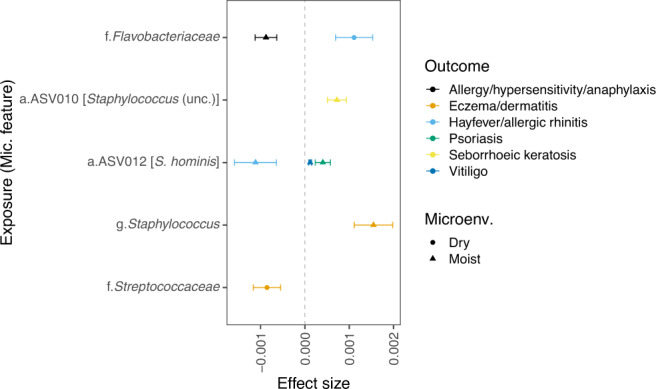
Despite the increasing knowledge about factors shaping the human microbiome, the host genetic factors that modulate the skin-microbiome interactions are still largely understudied. This contrasts with recent efforts to characterize host genes that influence the gut microbiota. Here, we investigated the effect of genetics on skin microbiota across three different skin microenvironments through meta-analyses of genome-wide association studies (GWAS) of two population-based German cohorts. We identified 23 genome-wide significant loci harboring 30 candidate genes involved in innate immune signaling, environmental sensing, cell differentiation, proliferation and fibroblast activity. However, no locus passed the strict threshold for study-wide significance (P < 6.3 × 10-10 for 80 features included in the analysis). Mendelian randomization (MR) analysis indicated the influence of staphylococci on eczema/dermatitis and suggested modulating effects of the microbiota on other skin diseases. Finally, transcriptional profiles of keratinocytes significantly changed after in vitro co-culturing with Staphylococcus epidermidis, chosen as a representative of skin commensals. Seven candidate genes from the GWAS were found overlapping with differential expression in the co-culturing experiments, warranting further research of the skin commensal and host genetic makeup interaction.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Moitinho-Silva, L. et al. Host traits, lifestyle and environment are associated with the human skin bacteria. Br J Dermatol, 10.1111/bjd.20072 (2021). - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
