

Semi-automated assembly of high-quality diploid human reference genomes
- PMID: 36261518
- PMCID: PMC9668749
- DOI: 10.1038/s41586-022-05325-5
Semi-automated assembly of high-quality diploid human reference genomes
Abstract
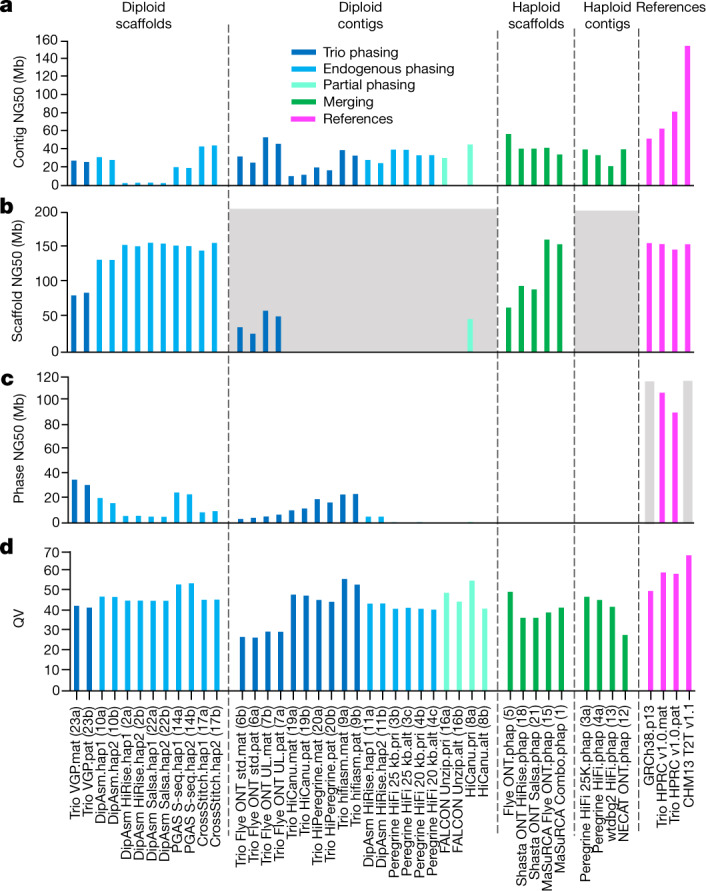
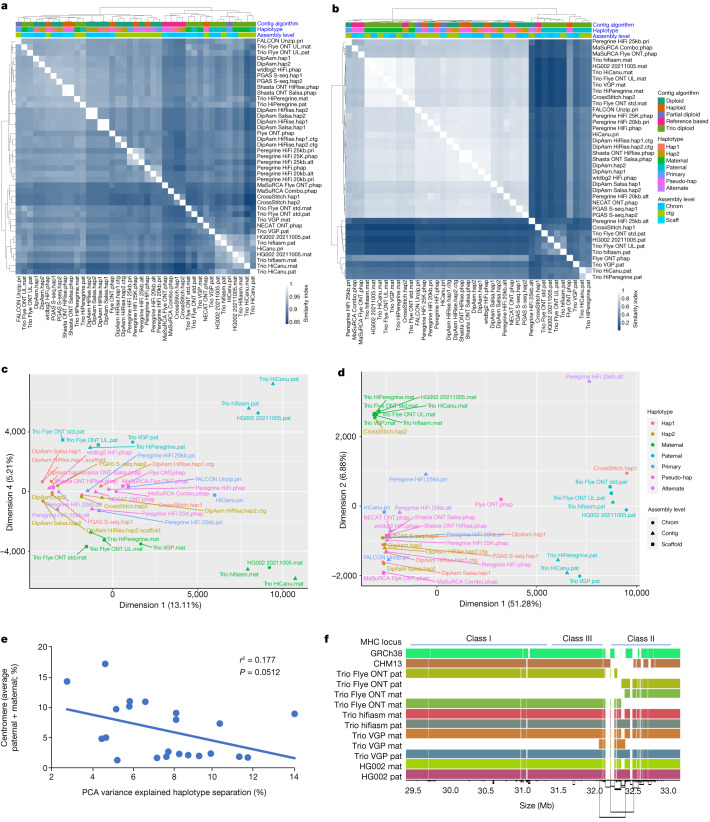
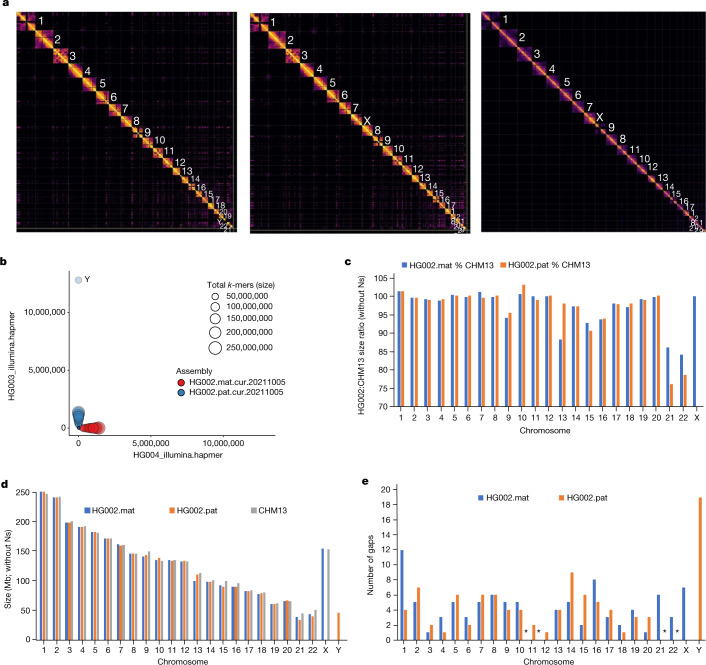
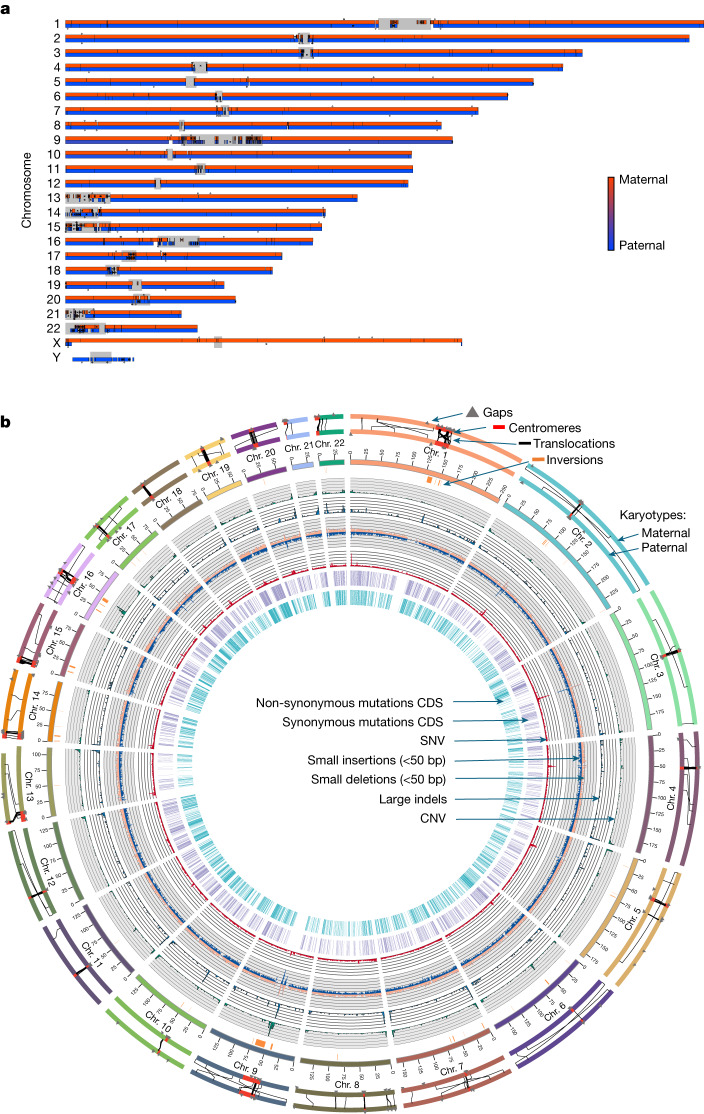
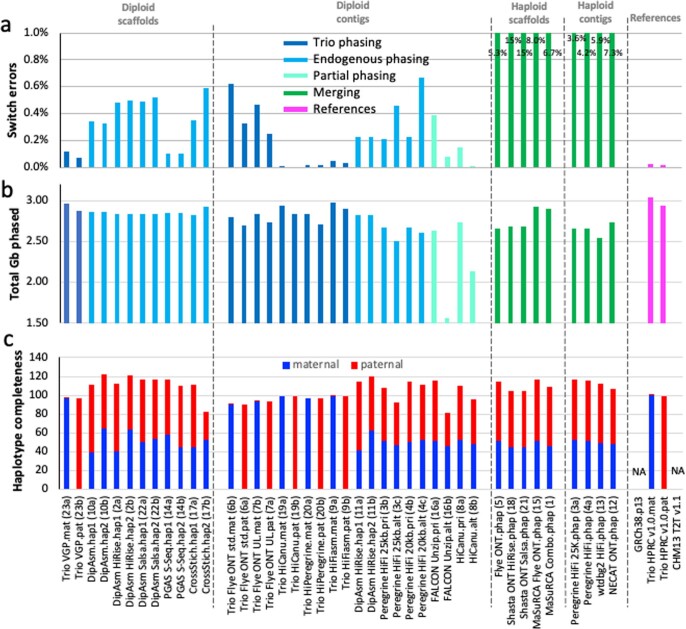
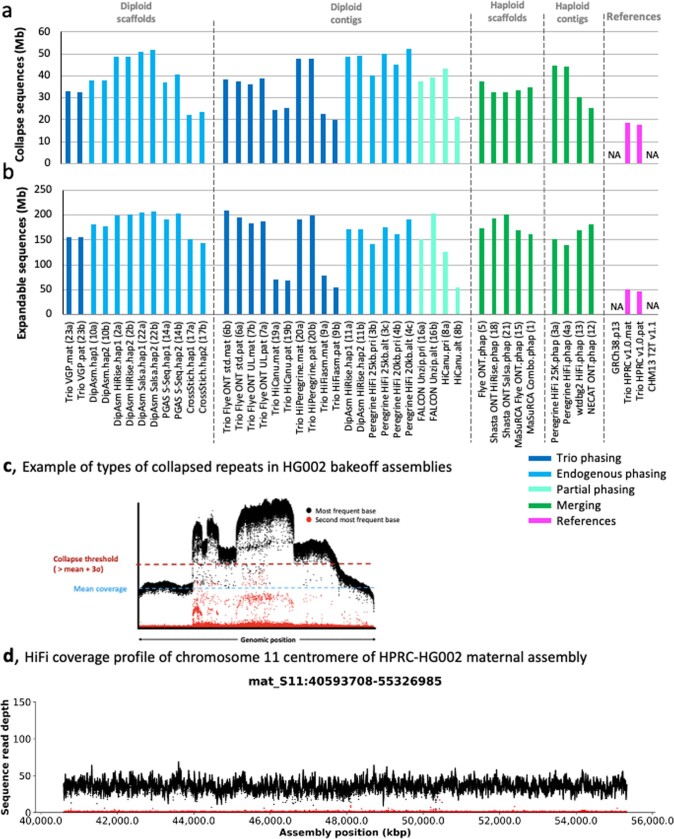
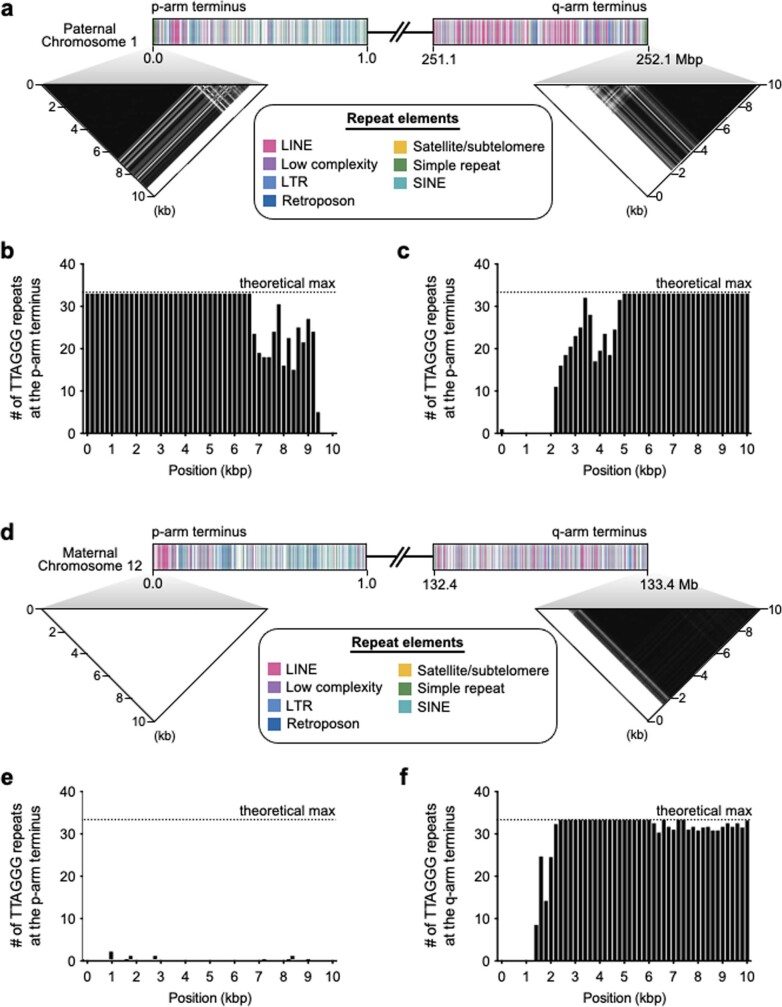
The current human reference genome, GRCh38, represents over 20 years of effort to generate a high-quality assembly, which has benefitted society1,2. However, it still has many gaps and errors, and does not represent a biological genome as it is a blend of multiple individuals3,4. Recently, a high-quality telomere-to-telomere reference, CHM13, was generated with the latest long-read technologies, but it was derived from a hydatidiform mole cell line with a nearly homozygous genome5. To address these limitations, the Human Pangenome Reference Consortium formed with the goal of creating high-quality, cost-effective, diploid genome assemblies for a pangenome reference that represents human genetic diversity6. Here, in our first scientific report, we determined which combination of current genome sequencing and assembly approaches yield the most complete and accurate diploid genome assembly with minimal manual curation. Approaches that used highly accurate long reads and parent-child data with graph-based haplotype phasing during assembly outperformed those that did not. Developing a combination of the top-performing methods, we generated our first high-quality diploid reference assembly, containing only approximately four gaps per chromosome on average, with most chromosomes within ±1% of the length of CHM13. Nearly 48% of protein-coding genes have non-synonymous amino acid changes between haplotypes, and centromeric regions showed the highest diversity. Our findings serve as a foundation for assembling near-complete diploid human genomes at scale for a pangenome reference to capture global genetic variation from single nucleotides to structural rearrangements.
© 2022. The Author(s).
Conflict of interest statement
E.E.E. was a scientific advisory board member of Variant Bio, Inc. J.K. and A.W. were full-time employees at Pacific Biosciences, a company developing single-molecule sequencing technologies. A.H. and J. Lee. were employees of Bionano Genomics, a company developing optical maps for genome assembly. J.G. and R.E.G. were affiliated with Dovetail Genomics, a company developing genome assembly tools, including Hi-C. A. Granat and E.B.J. were employees of Ilumina, Inc., a genome company generating short reads. A. Schmitt and S.S. were employees of Arima Genomics, a company developing Hi-C data for genome assemblies. All other authors declare no competing interests.
Figures
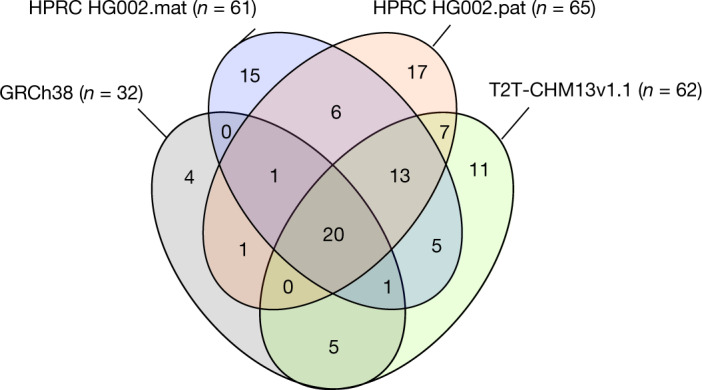
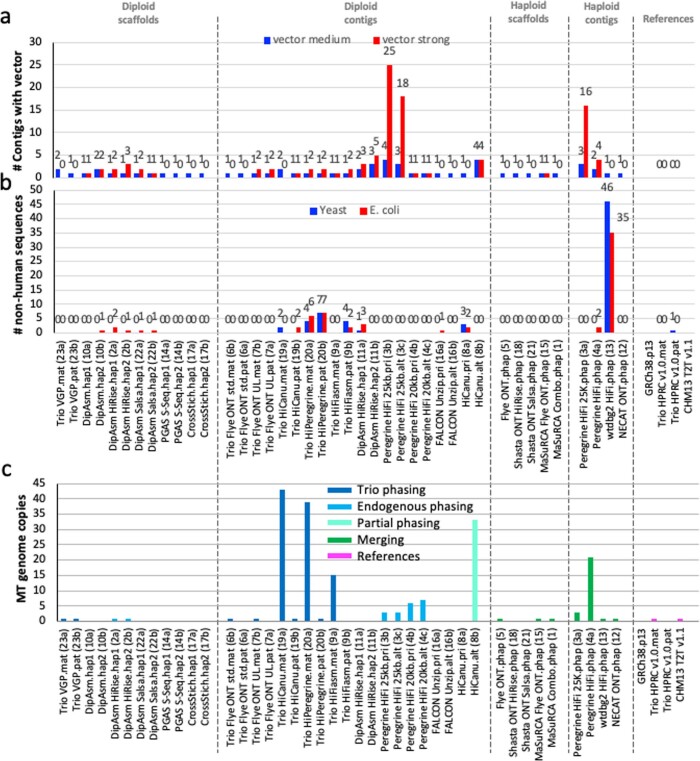
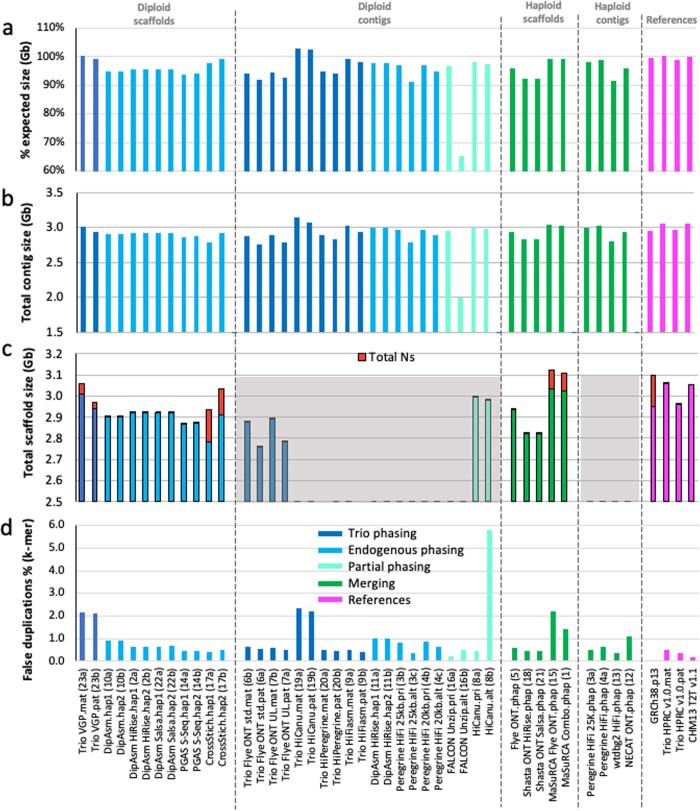
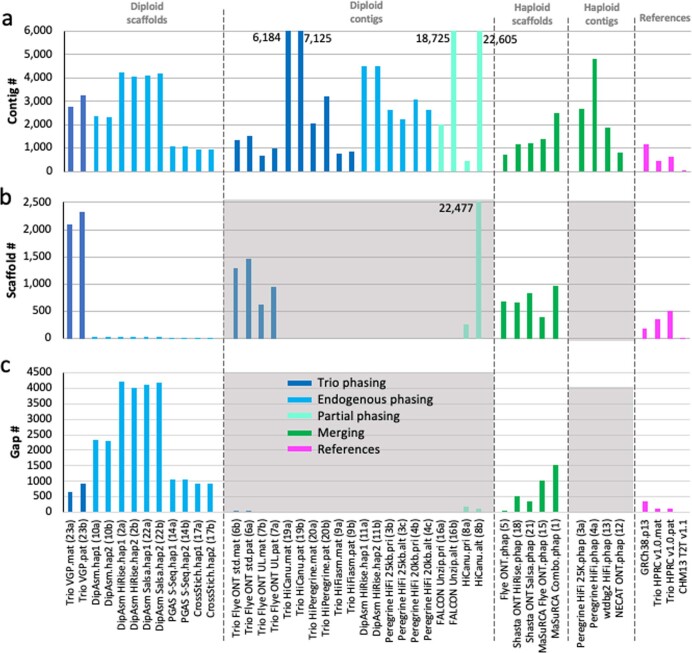
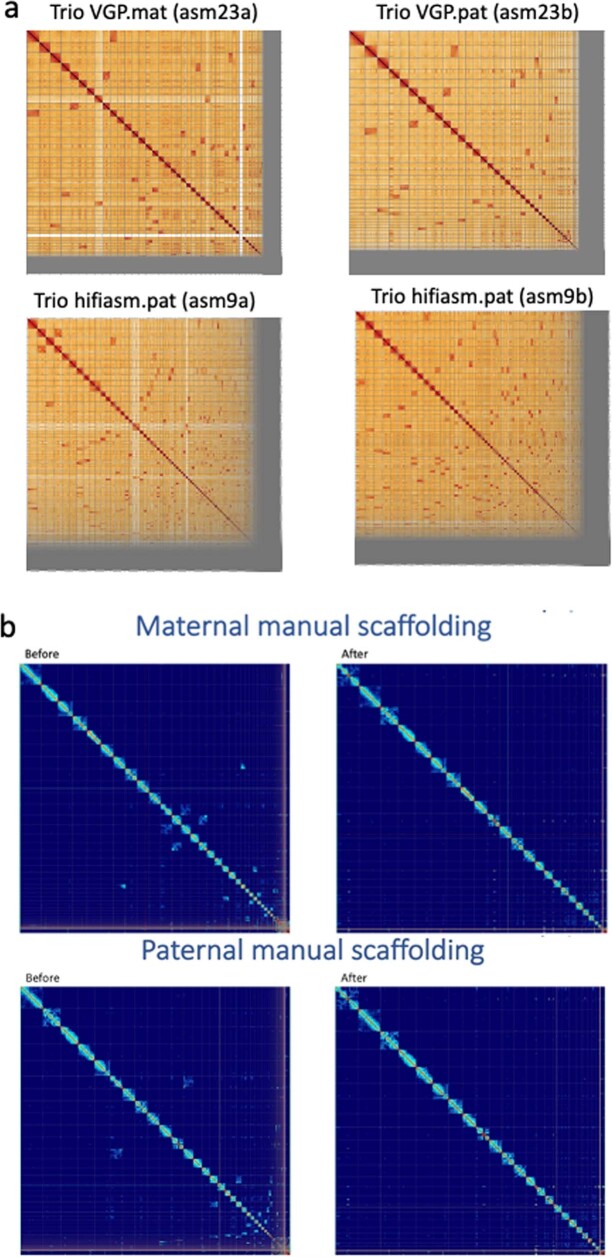
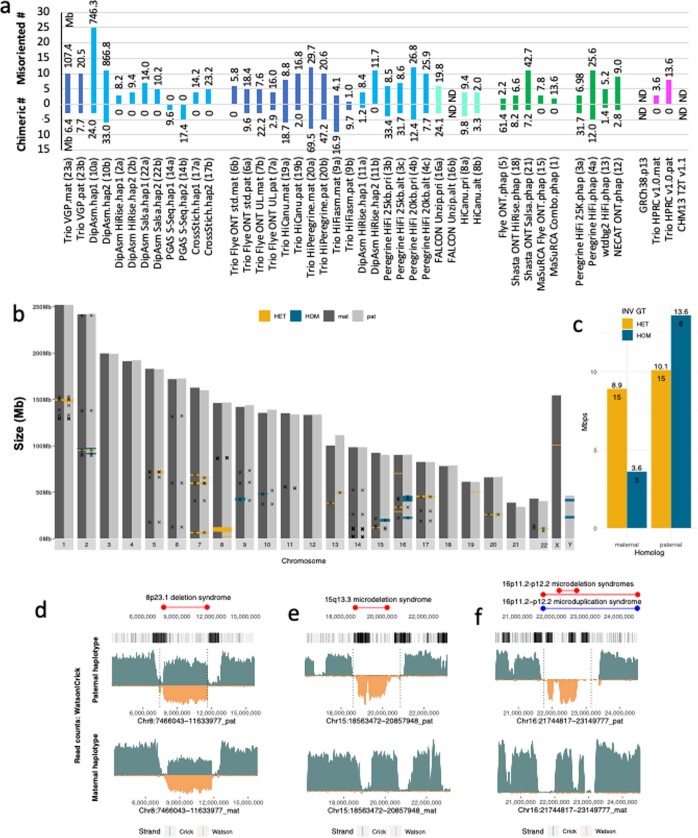
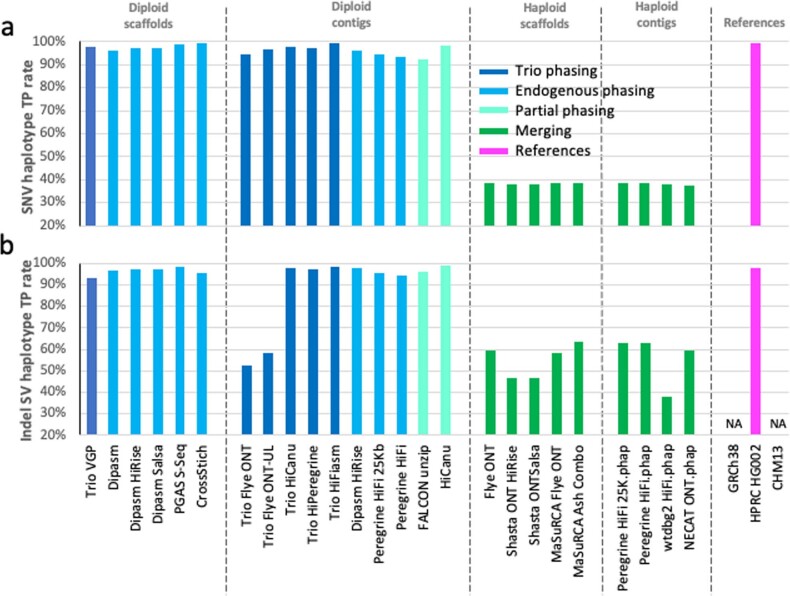
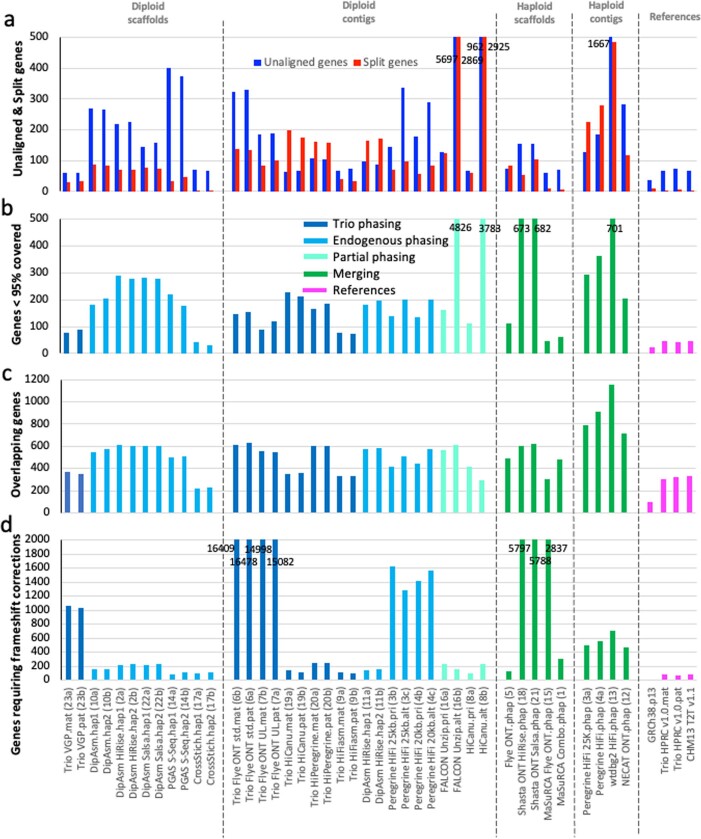

References
-
- Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
