

Evolution of immune genes is associated with the Black Death
- PMID: 36261521
- PMCID: PMC9580435
- DOI: 10.1038/s41586-022-05349-x
Evolution of immune genes is associated with the Black Death
Erratum in
-
Author Correction: Evolution of immune genes is associated with the Black Death.Nature. 2025 Jan;637(8048):E30. doi: 10.1038/s41586-024-08522-6. Nature. 2025. PMID: 39814901 Free PMC article. No abstract available.
Abstract
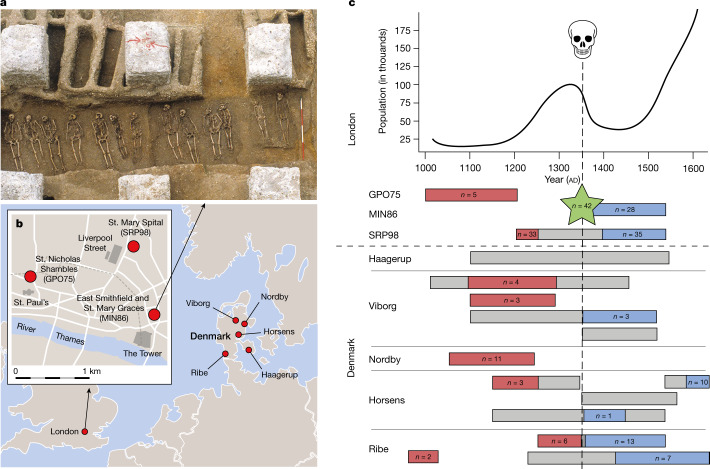
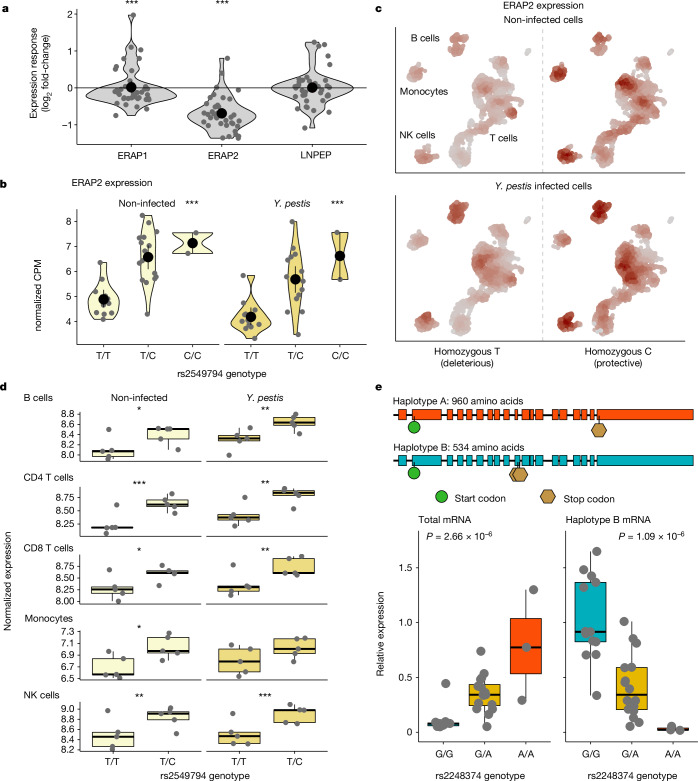
Infectious diseases are among the strongest selective pressures driving human evolution1,2. This includes the single greatest mortality event in recorded history, the first outbreak of the second pandemic of plague, commonly called the Black Death, which was caused by the bacterium Yersinia pestis3. This pandemic devastated Afro-Eurasia, killing up to 30-50% of the population4. To identify loci that may have been under selection during the Black Death, we characterized genetic variation around immune-related genes from 206 ancient DNA extracts, stemming from two different European populations before, during and after the Black Death. Immune loci are strongly enriched for highly differentiated sites relative to a set of non-immune loci, suggesting positive selection. We identify 245 variants that are highly differentiated within the London dataset, four of which were replicated in an independent cohort from Denmark, and represent the strongest candidates for positive selection. The selected allele for one of these variants, rs2549794, is associated with the production of a full-length (versus truncated) ERAP2 transcript, variation in cytokine response to Y. pestis and increased ability to control intracellular Y. pestis in macrophages. Finally, we show that protective variants overlap with alleles that are today associated with increased susceptibility to autoimmune diseases, providing empirical evidence for the role played by past pandemics in shaping present-day susceptibility to disease.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
J.K., A. Devault and J.-M.R. declare financial interest in Daicel Arbor Biosciences, which provided the myBaits hybridization capture kits for this work. All other authors declare no competing interests.
Figures












Comment in
-
Ancient DNA reveals rapid natural selection during the Black Death.Nature. 2022 Nov;611(7935):237-238. doi: 10.1038/d41586-022-03160-2. Nature. 2022. PMID: 36261712 No abstract available.
-
Uncovering the genomic toll of the Black Death.Trends Immunol. 2023 Feb;44(2):90-92. doi: 10.1016/j.it.2022.12.001. Epub 2022 Dec 14. Trends Immunol. 2023. PMID: 36526581
-
The Immune Ghosts of Pandemics Past.Gastroenterology. 2023 Apr;164(4):696. doi: 10.1053/j.gastro.2022.12.038. Epub 2023 Jan 3. Gastroenterology. 2023. PMID: 36608717 No abstract available.
-
Insufficient evidence for natural selection associated with the Black Death.Nature. 2025 Feb;638(8051):E19-E22. doi: 10.1038/s41586-024-08496-5. Epub 2025 Feb 19. Nature. 2025. PMID: 39972236 Free PMC article. No abstract available.
References
-
- Inhorn, M. C. & Brown, P. J. The anthropology of infectious disease. Anu. Rev. Anthrolpol.19, 89–117 (1990).
-
- Benedictow, O. J. The Black Death, 1346–1353: The Complete History (Boydell Press, 2004).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
