

An integrated transcriptome mapping the regulatory network of coding and long non-coding RNAs provides a genomics resource in chickpea
- PMID: 36261617
- PMCID: PMC9581958
- DOI: 10.1038/s42003-022-04083-4
An integrated transcriptome mapping the regulatory network of coding and long non-coding RNAs provides a genomics resource in chickpea
Abstract
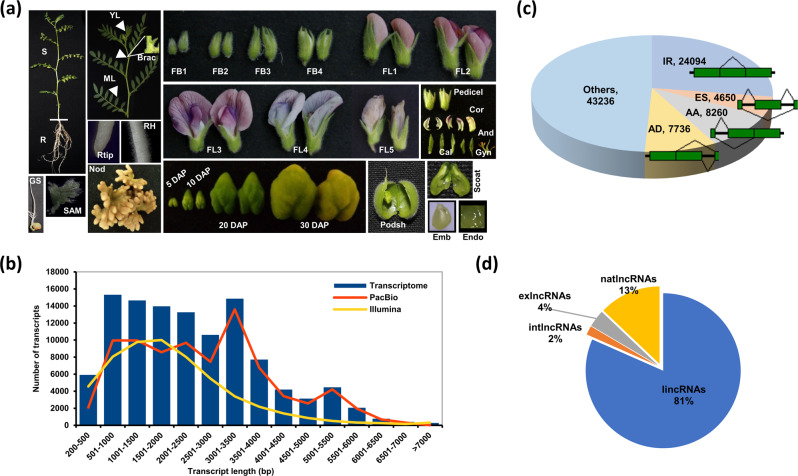
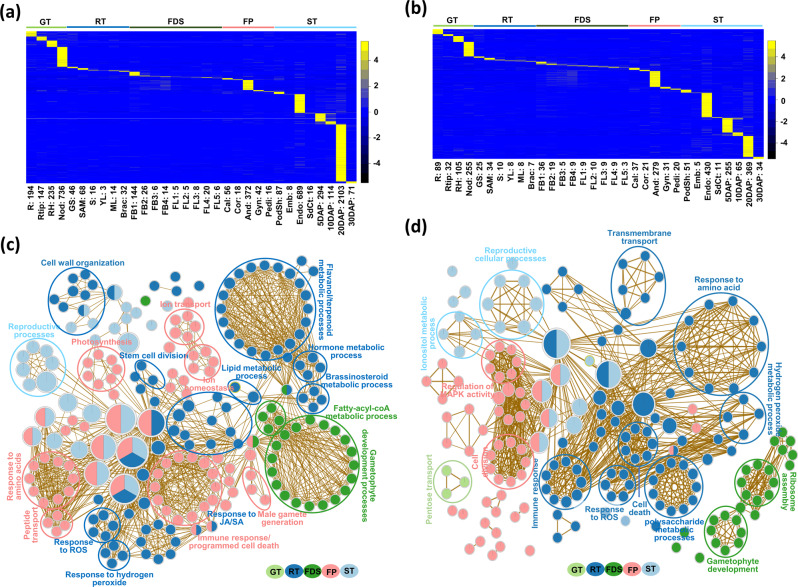
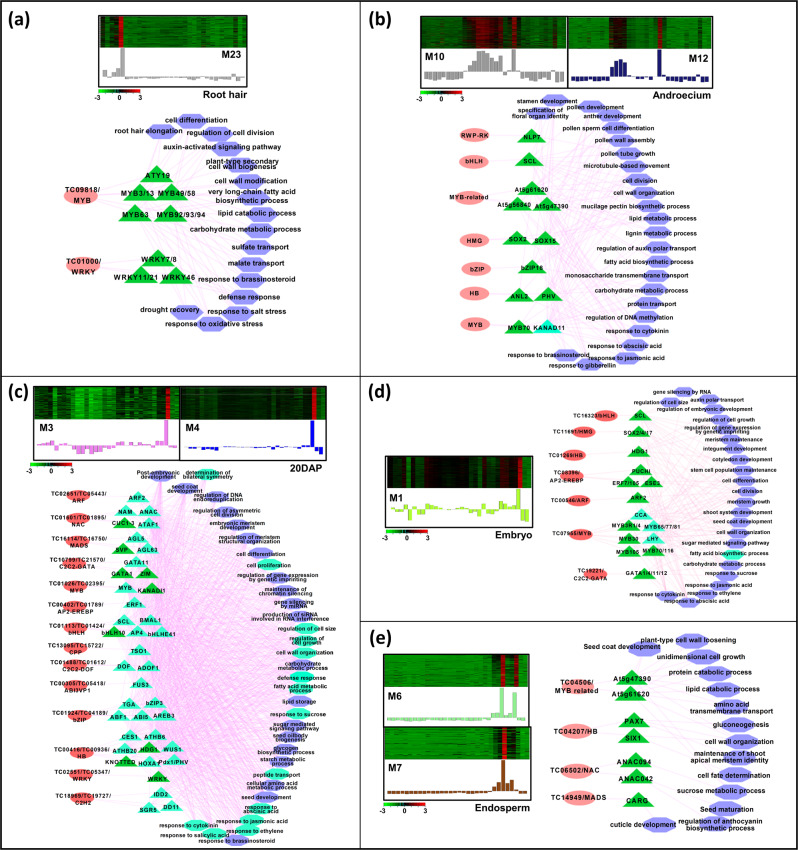
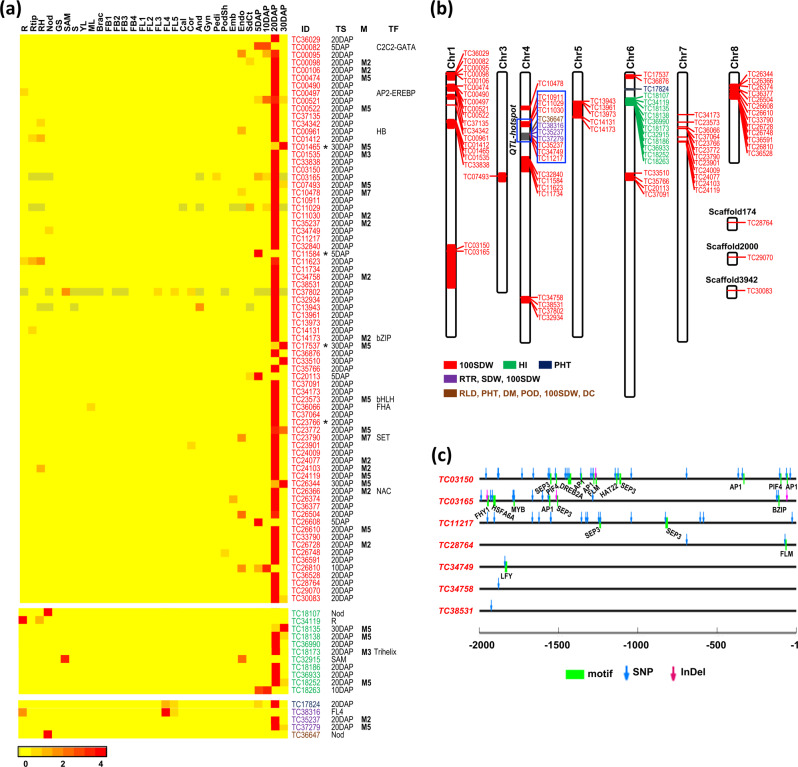
Large-scale transcriptome analysis can provide a systems-level understanding of biological processes. To accelerate functional genomic studies in chickpea, we perform a comprehensive transcriptome analysis to generate full-length transcriptome and expression atlas of protein-coding genes (PCGs) and long non-coding RNAs (lncRNAs) from 32 different tissues/organs via deep sequencing. The high-depth RNA-seq dataset reveal expression dynamics and tissue-specificity along with associated biological functions of PCGs and lncRNAs during development. The coexpression network analysis reveal modules associated with a particular tissue or a set of related tissues. The components of transcriptional regulatory networks (TRNs), including transcription factors, their cognate cis-regulatory motifs, and target PCGs/lncRNAs that determine developmental programs of different tissues/organs, are identified. Several candidate tissue-specific and abiotic stress-responsive transcripts associated with quantitative trait loci that determine important agronomic traits are also identified. These results provide an important resource to advance functional/translational genomic and genetic studies during chickpea development and environmental conditions.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



Similar articles
-
Genome-wide analysis of long intergenic non-coding RNAs in chickpea and their potential role in flower development.Sci Rep. 2016 Sep 15;6:33297. doi: 10.1038/srep33297. Sci Rep. 2016. PMID: 27628568 Free PMC article.
-
Comprehensive transcriptomic analysis of two RIL parents with contrasting salt responsiveness identifies polyadenylated and non-polyadenylated flower lncRNAs in chickpea.Plant Biotechnol J. 2022 Jul;20(7):1402-1416. doi: 10.1111/pbi.13822. Epub 2022 May 13. Plant Biotechnol J. 2022. PMID: 35395125 Free PMC article.
-
Genome-wide analysis suggests the potential role of lncRNAs during seed development and seed size/weight determination in chickpea.Planta. 2022 Sep 12;256(4):79. doi: 10.1007/s00425-022-03986-0. Planta. 2022. PMID: 36094579
-
Current advances in chickpea genomics: applications and future perspectives.Plant Cell Rep. 2018 Jul;37(7):947-965. doi: 10.1007/s00299-018-2305-6. Epub 2018 Jun 2. Plant Cell Rep. 2018. PMID: 29860584 Review.
-
The Working Modules of Long Noncoding RNAs in Cancer Cells.Adv Exp Med Biol. 2016;927:49-67. doi: 10.1007/978-981-10-1498-7_2. Adv Exp Med Biol. 2016. PMID: 27376731 Review.
Cited by
-
Identification and expression of the AREB/ABF/ABI5 subfamily genes in chickpea and lentil reveal major players involved in ABA-mediated defense response to drought stress.Planta. 2025 Jun 10;262(1):22. doi: 10.1007/s00425-025-04740-y. Planta. 2025. PMID: 40493071
-
Phenotypic and genetic characterization of a near-isogenic line pair: insights into flowering time in chickpea.BMC Plant Biol. 2024 Jul 25;24(1):709. doi: 10.1186/s12870-024-05411-y. BMC Plant Biol. 2024. PMID: 39054447 Free PMC article.
-
Insights into Bactericera cockerelli and Candidatus Liberibacter solanacearum interaction: a tissue-specific transcriptomic approach.Front Plant Sci. 2024 Aug 30;15:1393994. doi: 10.3389/fpls.2024.1393994. eCollection 2024. Front Plant Sci. 2024. PMID: 39280947 Free PMC article.
-
Functional Genomics: From Soybean to Legume.Int J Mol Sci. 2025 Jun 30;26(13):6323. doi: 10.3390/ijms26136323. Int J Mol Sci. 2025. PMID: 40650105 Free PMC article. Review.
-
Unravelling Differential DNA Methylation Patterns in Genotype Dependent Manner under Salinity Stress Response in Chickpea.Int J Mol Sci. 2023 Jan 18;24(3):1863. doi: 10.3390/ijms24031863. Int J Mol Sci. 2023. PMID: 36768187 Free PMC article.
References
-
- Tiwari BK, Tripathi PN. Yield gap analysis of chickpea (Cicer arietinum) through front line demonstration on farmer’s fields. J. Rural Agric. Res. 2014;14:5–8.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
