

This is a preprint.
A pseudovirus system enables deep mutational scanning of the full SARS-CoV-2 spike
- PMID: 36263061
- PMCID: PMC9580381
- DOI: 10.1101/2022.10.13.512056
A pseudovirus system enables deep mutational scanning of the full SARS-CoV-2 spike
Update in
-
A pseudovirus system enables deep mutational scanning of the full SARS-CoV-2 spike.Cell. 2023 Mar 16;186(6):1263-1278.e20. doi: 10.1016/j.cell.2023.02.001. Epub 2023 Feb 13. Cell. 2023. PMID: 36868218 Free PMC article.
Abstract
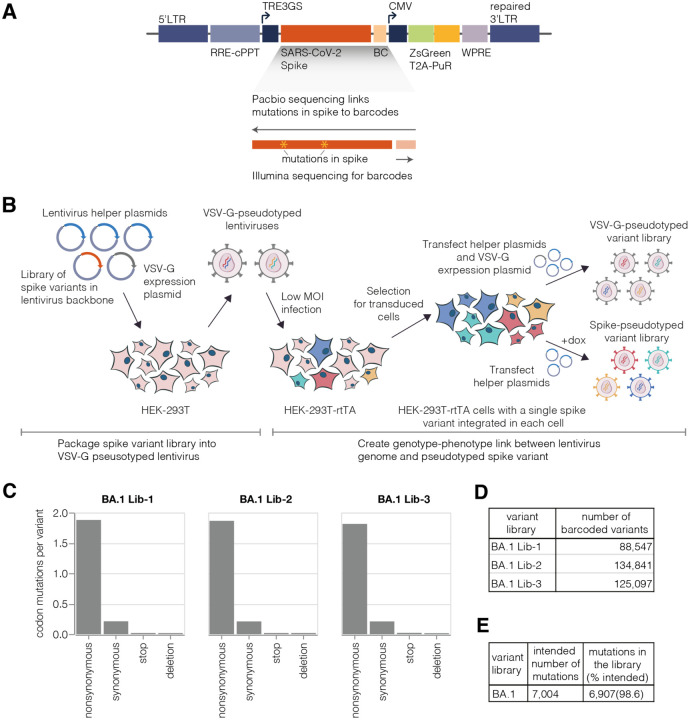
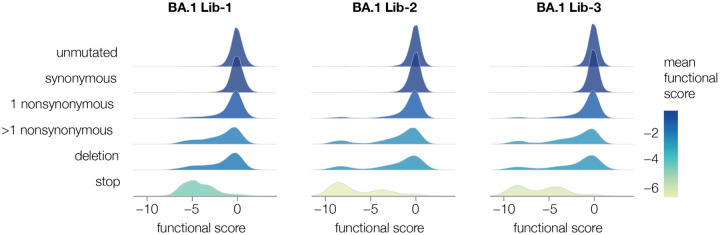
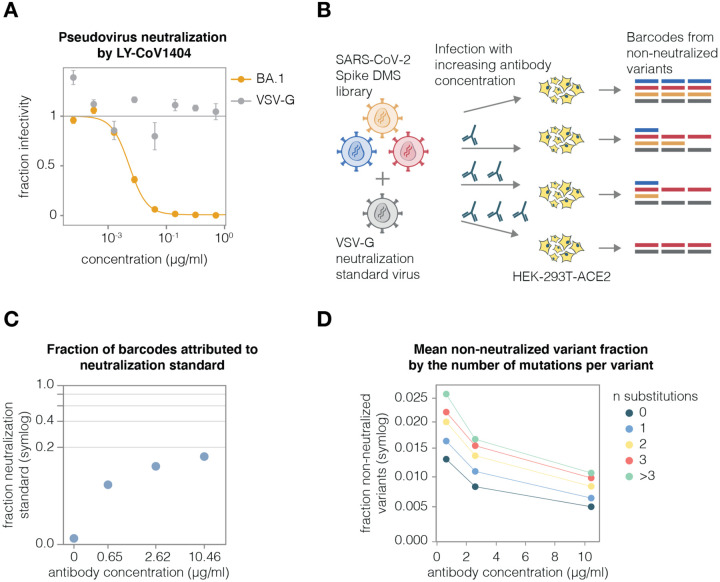
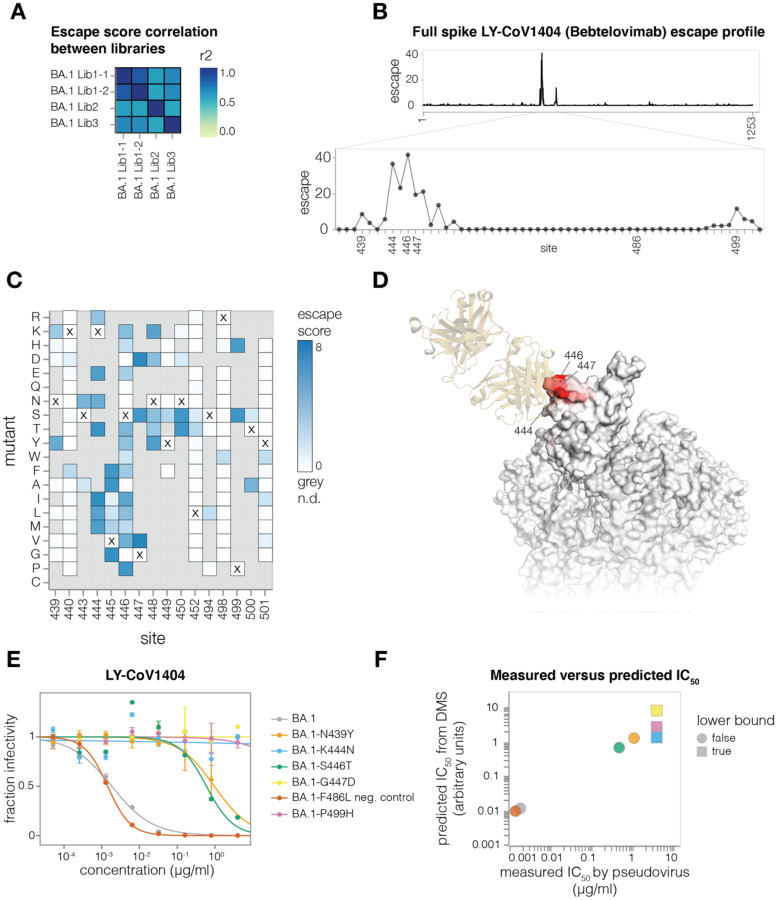
A major challenge in understanding SARS-CoV-2 evolution is interpreting the antigenic and functional effects of emerging mutations in the viral spike protein. Here we describe a new deep mutational scanning platform based on non-replicative pseudotyped lentiviruses that directly quantifies how large numbers of spike mutations impact antibody neutralization and pseudovirus infection. We demonstrate this new platform by making libraries of the Omicron BA.1 and Delta spikes. These libraries each contain ~7000 distinct amino-acid mutations in the context of up to ~135,000 unique mutation combinations. We use these libraries to map escape mutations from neutralizing antibodies targeting the receptor binding domain, N-terminal domain, and S2 subunit of spike. Overall, this work establishes a high-throughput and safe approach to measure how ~10 5 combinations of mutations affect antibody neutralization and spike-mediated infection. Notably, the platform described here can be extended to the entry proteins of many other viruses.
Conflict of interest statement
Competing interests
JDB is on the scientific advisory board of Apriori Bio and Oncorus, and has recently consulted on topics related to viral evolution for Moderna and Merck. JDB, KHDC, and CER receive royalty payments as inventors on Fred Hutch licensed patents related to viral deep mutational scanning. JDB, KHDC, CER and BD are inventors on a pending patent application relating to the viral deep mutational scanning system described in this paper. RB is a consultant for IAVI, Adagio, Adimab, Mabloc, VosBio, Nonigenex, and Radiant. DDH is a co-founder of TaiMed Biologics and RenBio, consultant to WuXi Biologics and Brii Biosciences, and board director for Vicarious Surgical. The other authors declare no competing interests.
Figures
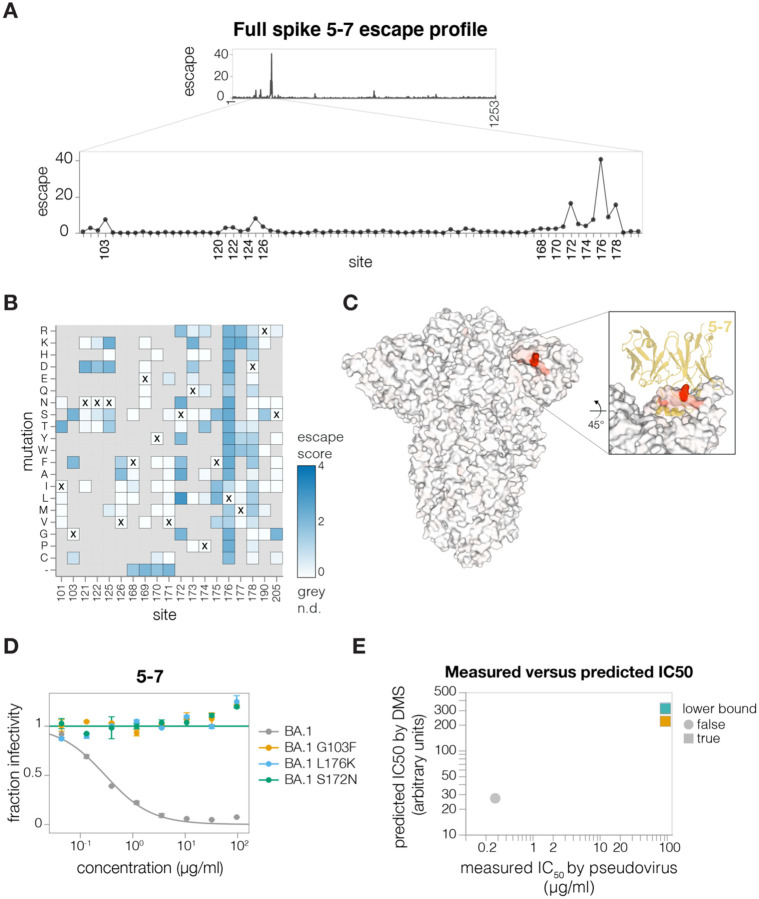

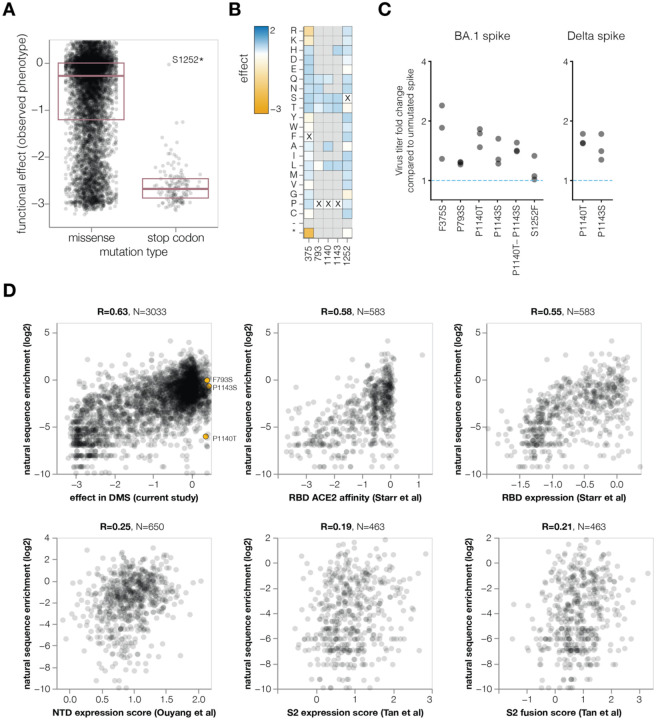
References
-
- Ballal A., Malliaris C.D., Morozov A.V., 2020. Molecular Phenotypes as Key Intermediates in Mapping Genotypes to Fitness, in: Pontarotti P. (Ed.), Evolutionary Biology—A Transdisciplinary Approach. Springer International Publishing, Cham, pp. 15–40. 10.1007/978-3-030-57246-4_2 - DOI
-
- Baum A., Fulton B.O., Wloga E., Copin R., Pascal K.E., Russo V., Giordano S., Lanza K., Negron N., Ni M., Wei Y., Atwal G.S., Murphy A.J., Stahl N., Yancopoulos G.D., Kyratsous C.A., 2020. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science eabd0831. 10.1126/science.abd0831 - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous