

A Molecular Switch between Mammalian MLL Complexes Dictates Response to Menin-MLL Inhibition
- PMID: 36264143
- PMCID: PMC9827117
- DOI: 10.1158/2159-8290.CD-22-0416
A Molecular Switch between Mammalian MLL Complexes Dictates Response to Menin-MLL Inhibition
Abstract
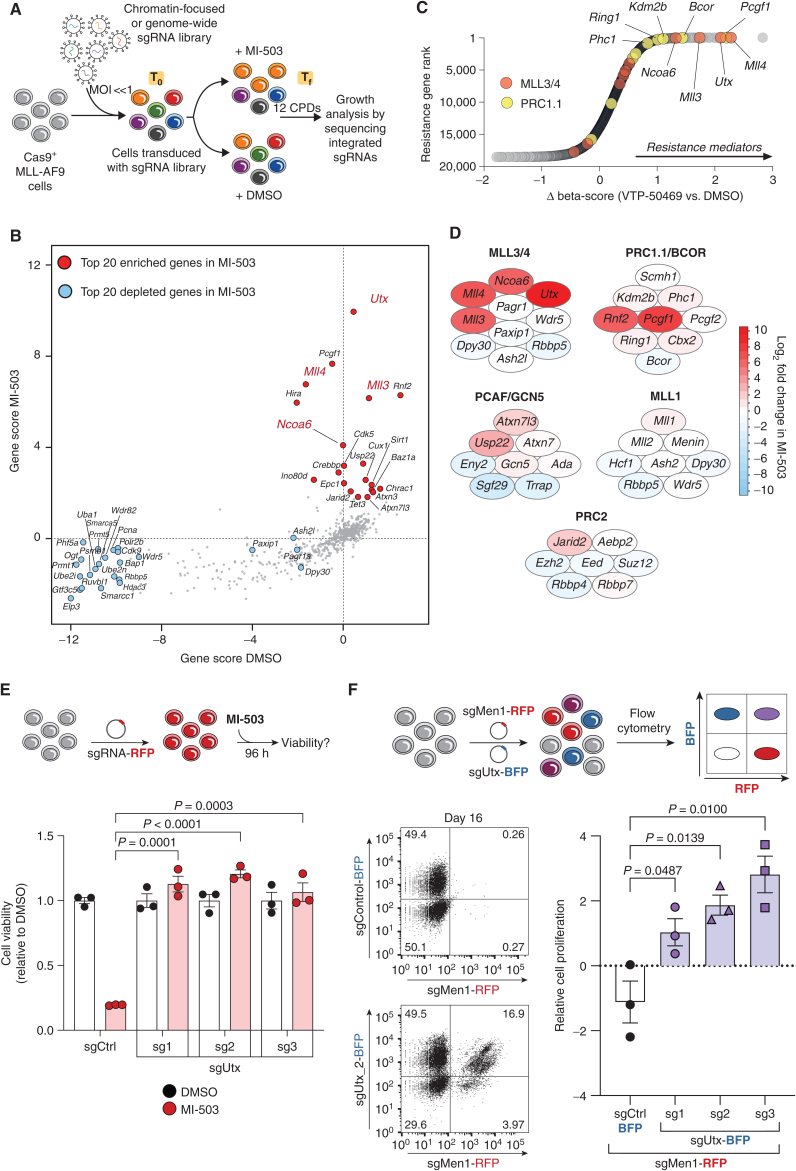
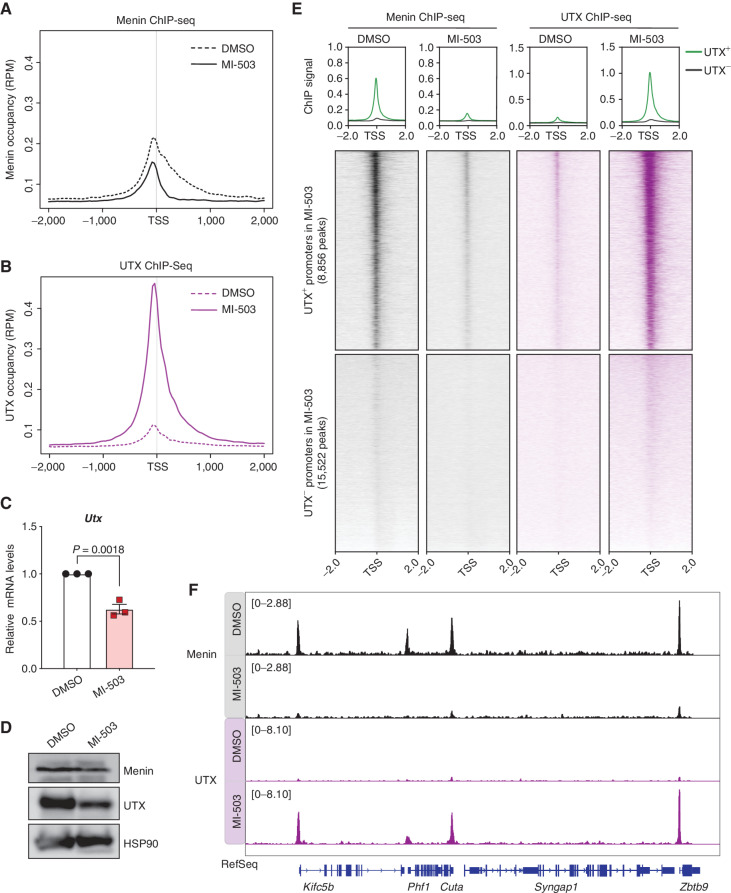
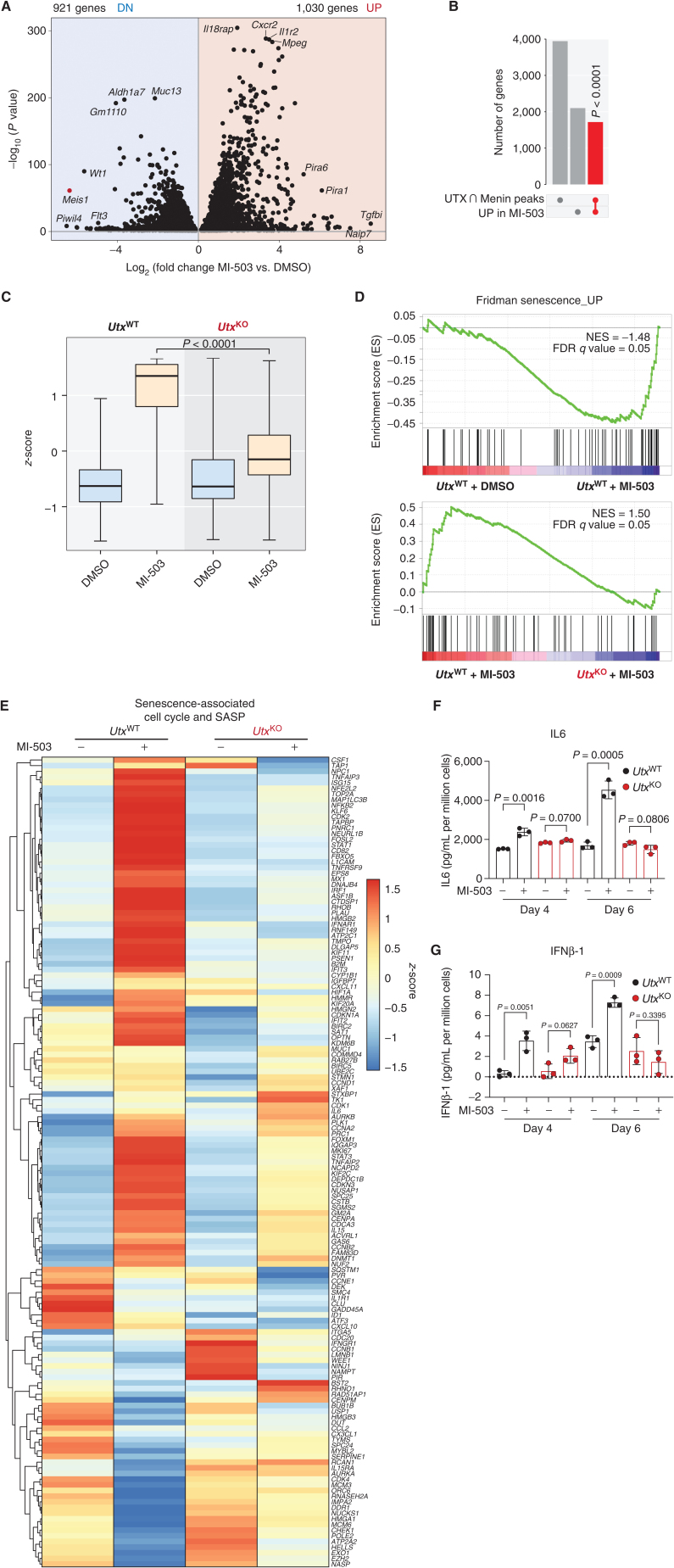
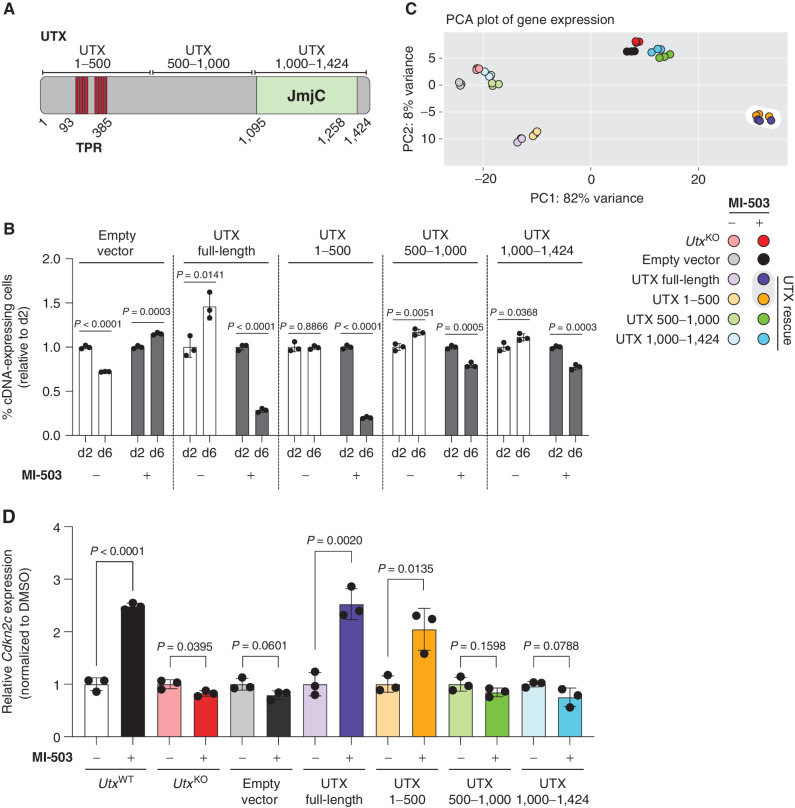
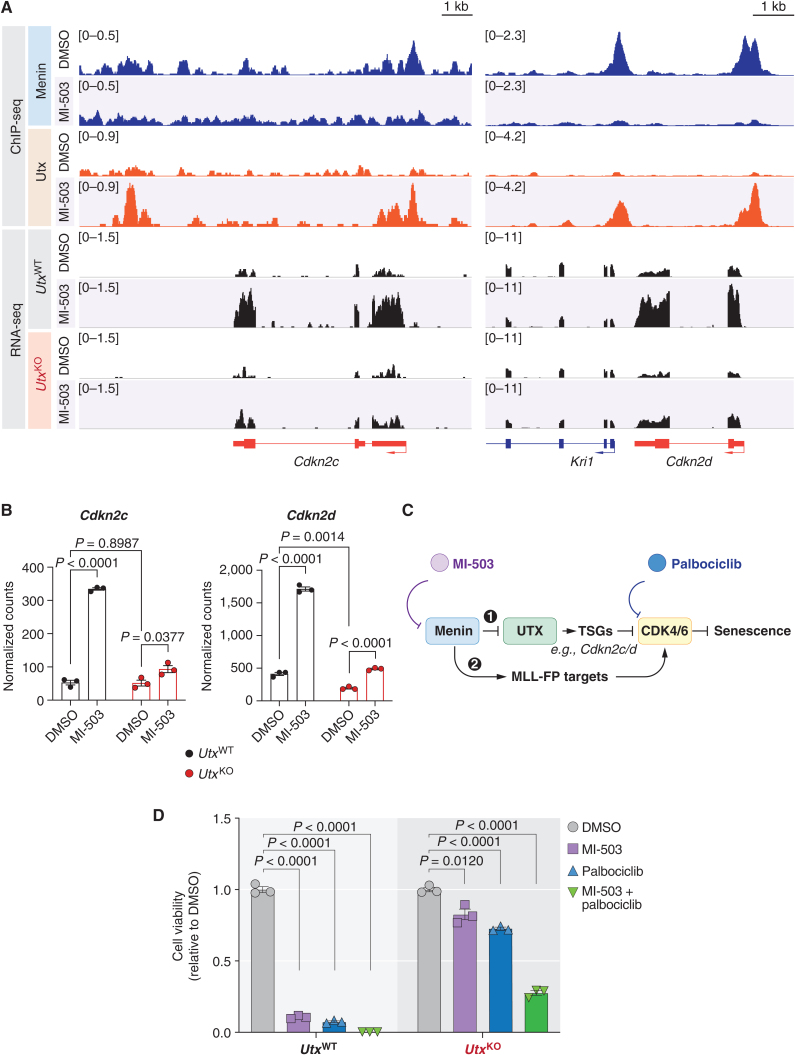
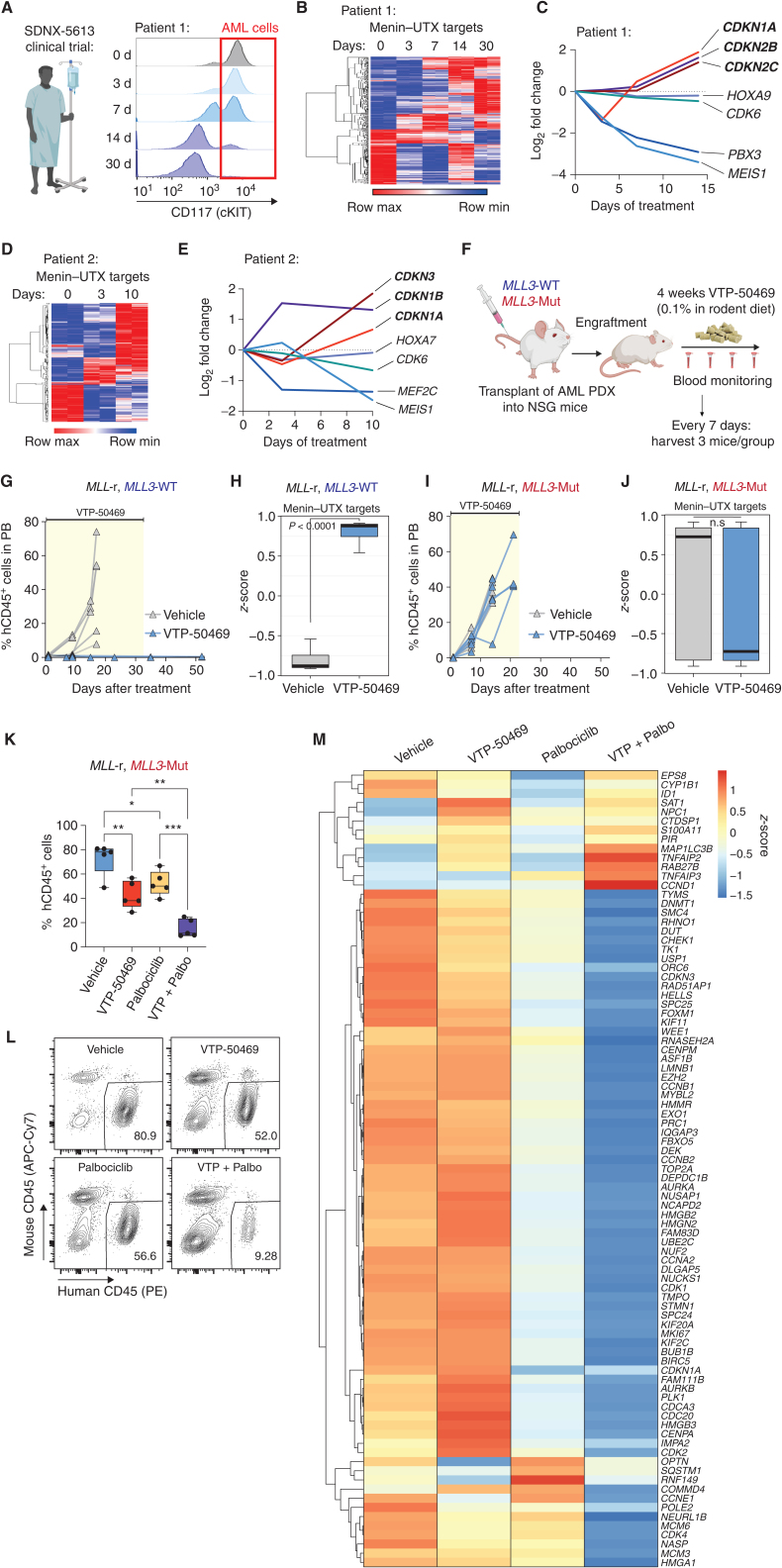
Menin interacts with oncogenic MLL1-fusion proteins, and small molecules that disrupt these associations are in clinical trials for leukemia treatment. By integrating chromatin-focused and genome-wide CRISPR screens with genetic, pharmacologic, and biochemical approaches, we discovered a conserved molecular switch between the MLL1-Menin and MLL3/4-UTX chromatin-modifying complexes that dictates response to Menin-MLL inhibitors. MLL1-Menin safeguards leukemia survival by impeding the binding of the MLL3/4-UTX complex at a subset of target gene promoters. Disrupting the Menin-MLL1 interaction triggers UTX-dependent transcriptional activation of a tumor-suppressive program that dictates therapeutic responses in murine and human leukemia. Therapeutic reactivation of this program using CDK4/6 inhibitors mitigates treatment resistance in leukemia cells that are insensitive to Menin inhibitors. These findings shed light on novel functions of evolutionarily conserved epigenetic mediators like MLL1-Menin and MLL3/4-UTX and are relevant to understand and target molecular pathways determining therapeutic responses in ongoing clinical trials.
Significance: Menin-MLL inhibitors silence a canonical HOX- and MEIS1-dependent oncogenic gene expression program in leukemia. We discovered a parallel, noncanonical transcriptional program involving tumor suppressor genes that are repressed in Menin-MLL inhibitor-resistant leukemia cells but that can be reactivated upon combinatorial treatment with CDK4/6 inhibitors to augment therapy responses. This article is highlighted in the In This Issue feature, p. 1.
©2022 The Authors; Published by the American Association for Cancer Research.
Figures

Comment in
- doi: 10.1158/2159-8290.CD-13-1-ITI
References
-
- Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell 2004;13:587–97. - PubMed
-
- Yokoyama A, Somervaille TCP, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 2005;123:207–18. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- T32 CA160001/CA/NCI NIH HHS/United States
- R01 GM040922/GM/NIGMS NIH HHS/United States
- K99 GM140265/GM/NIGMS NIH HHS/United States
- P50 CA206963/CA/NCI NIH HHS/United States
- P01 CA196539/CA/NCI NIH HHS/United States
- P50 CA254838/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- F32 CA217068/CA/NCI NIH HHS/United States
- R01 CA176745/CA/NCI NIH HHS/United States
- P01 CA013106/CA/NCI NIH HHS/United States
- R00 GM140265/GM/NIGMS NIH HHS/United States
- F31 CA192835/CA/NCI NIH HHS/United States
- R01 CA190261/CA/NCI NIH HHS/United States
- R01 CA204639/CA/NCI NIH HHS/United States
- P01 CA066996/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Research Materials
