

Prevalence and Disease Expression of Pathogenic and Likely Pathogenic Variants Associated With Inherited Cardiomyopathies in the General Population
- PMID: 36264615
- PMCID: PMC9770140
- DOI: 10.1161/CIRCGEN.122.003704
Prevalence and Disease Expression of Pathogenic and Likely Pathogenic Variants Associated With Inherited Cardiomyopathies in the General Population
Abstract
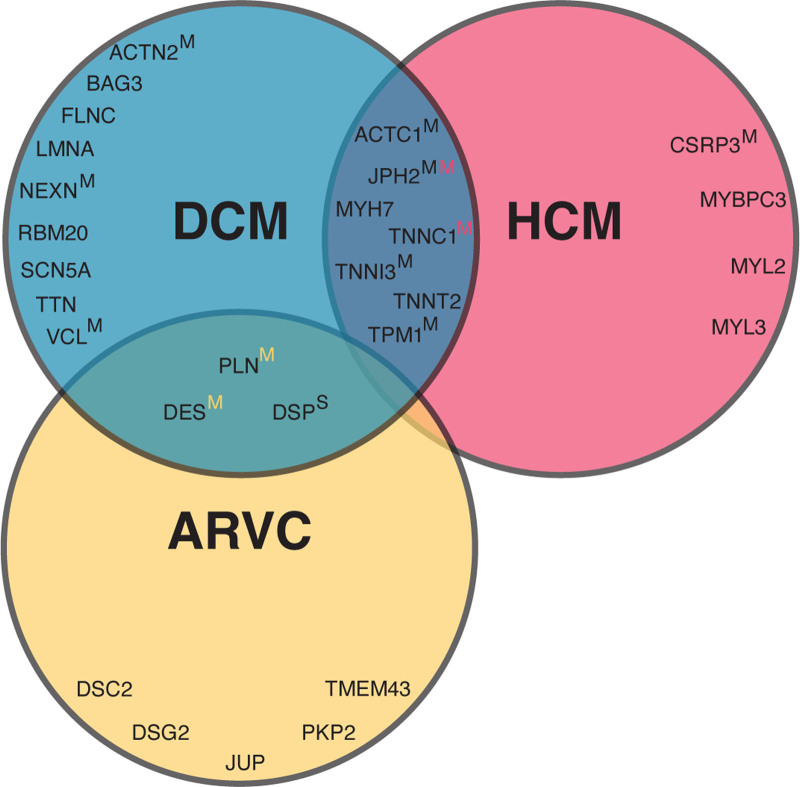
Background: Pathogenic and likely pathogenic variants associated with arrhythmogenic right ventricular cardiomyopathy (ARVC), dilated cardiomyopathy (DCM), and hypertrophic cardiomyopathy (HCM) are recommended to be reported as secondary findings in genome sequencing studies. This provides opportunities for early diagnosis, but also fuels uncertainty in variant carriers (G+), since disease penetrance is incomplete. We assessed the prevalence and disease expression of G+ in the general population.
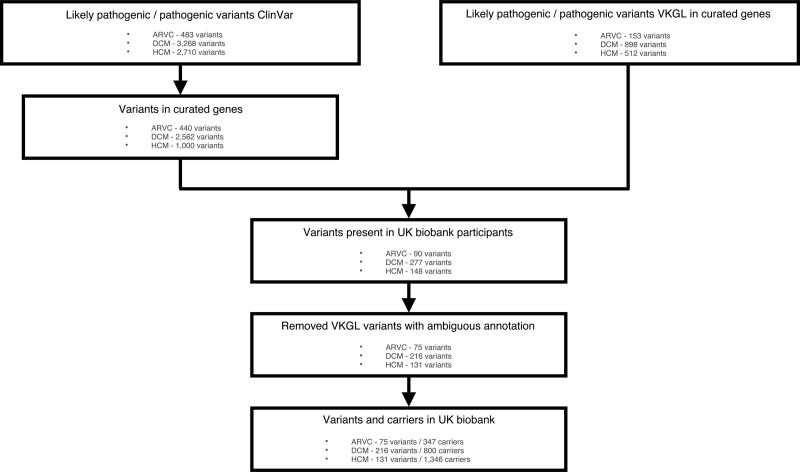
Methods: We identified pathogenic and likely pathogenic variants associated with ARVC, DCM and/or HCM in 200 643 UK Biobank individuals, who underwent whole exome sequencing. We calculated the prevalence of G+ and analyzed the frequency of cardiomyopathy/heart failure diagnosis. In undiagnosed individuals, we analyzed early signs of disease expression using available electrocardiography and cardiac magnetic resonance imaging data.
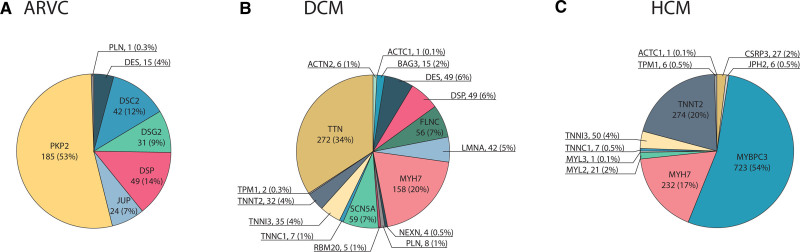
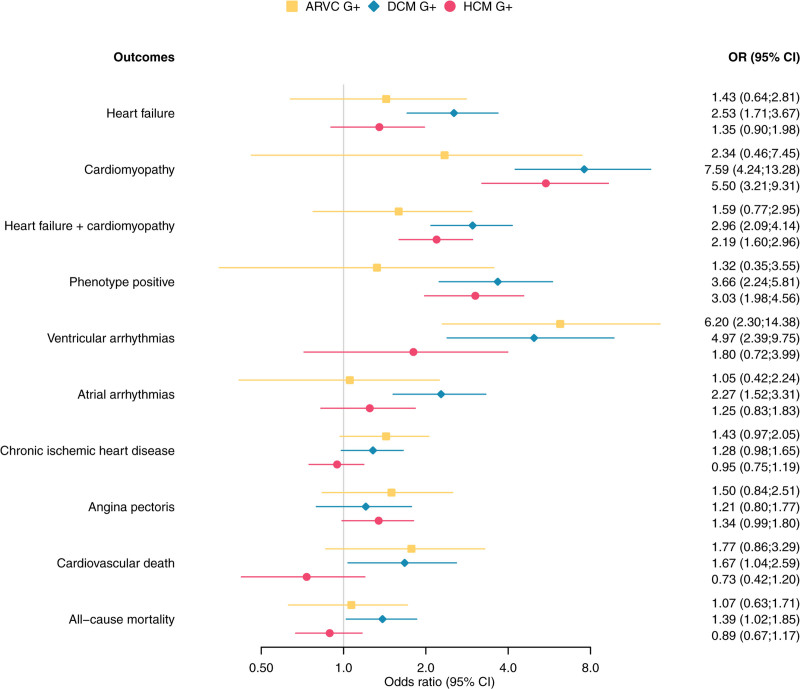
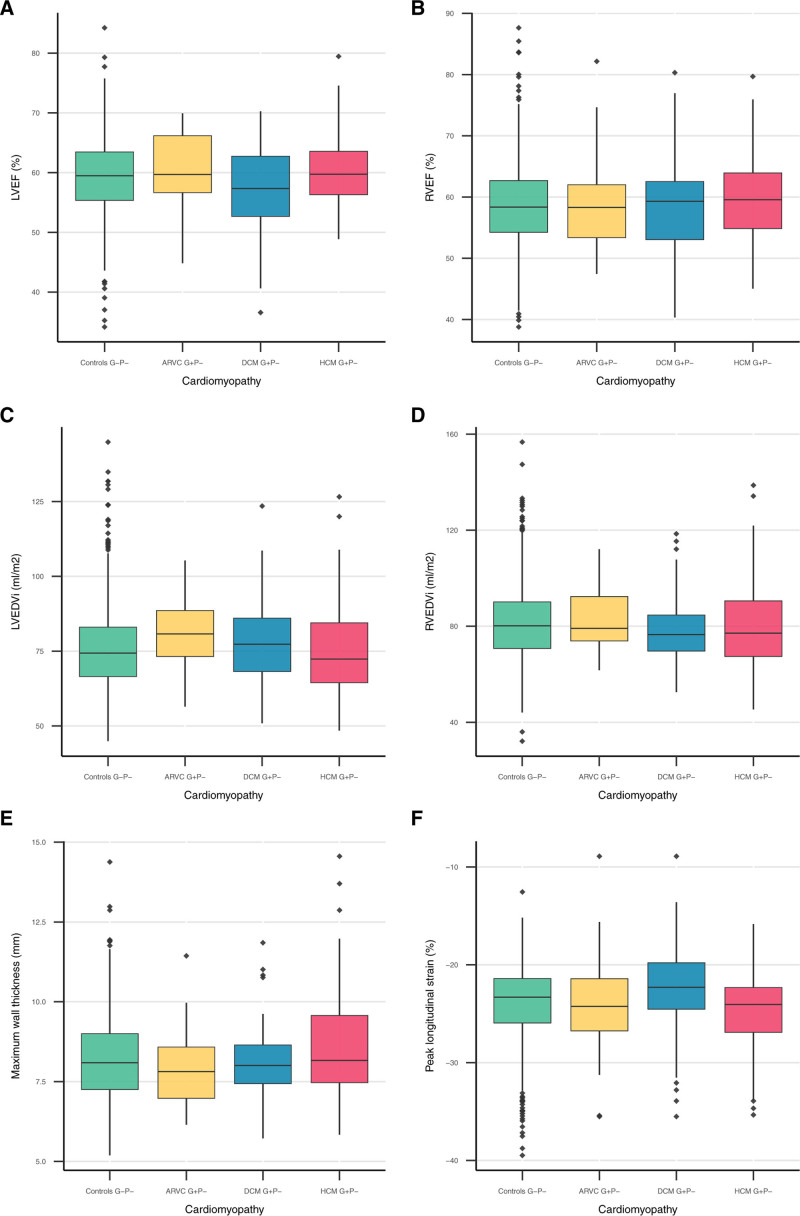
Results: We found a prevalence of 1:578, 1:251, and 1:149 for pathogenic and likely pathogenic variants associated with ARVC, DCM and HCM respectively. Compared with controls, cardiovascular mortality was higher in DCM G+ (odds ratio 1.67 [95% CI 1.04; 2.59], P=0.030), but similar in ARVC and HCM G+ (P≥0.100). Cardiomyopathy or heart failure diagnosis were more frequent in DCM G+ (odds ratio 3.66 [95% CI 2.24; 5.81], P=4.9×10-7) and HCM G+ (odds ratio 3.03 [95% CI 1.98; 4.56], P=5.8×10-7), but comparable in ARVC G+ (P=0.172). In contrast, ARVC G+ had more ventricular arrhythmias (P=3.3×10-4). In undiagnosed individuals, left ventricular ejection fraction was reduced in DCM G+ (P=0.009).
Conclusions: In the general population, pathogenic and likely pathogenic variants associated with ARVC, DCM, or HCM are not uncommon. Although G+ have increased mortality and morbidity, disease penetrance in these carriers from the general population remains low (1.2-3.1%). Follow-up decisions in case of incidental findings should not be based solely on a variant, but on multiple factors, including family history and disease expression.
Keywords: arrhythmogenic right ventricular cardiomyopathy; dilated cardiomyopathy; genetics; hypertrophic cardiomyopathy; whole exome sequencing.
Figures
References
-
- McKenna WJ, Judge DP. Epidemiology of the inherited cardiomyopathies. Nat Rev Cardiol. 2021;18:22–36. doi: 10.1038/s41569-020-0428-2 - PubMed
-
- Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10:531–547. doi: 10.1038/nrcardio.2013.105 - PubMed
-
- Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;124:2761–2796. doi: 10.1161/CIR.0b013e318223e230 - PubMed
-
- Miller DT, Lee K, Chung WK, Gordon AS, Herman GE, Klein TE, Stewart DR, Amendola LM, Adelman K, Bale SJ, et al. ACMG SF v3. 0 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021;23:1381–1390. doi: 10.1038/s41436-021-01172-3 - PubMed
-
- James CA, Jongbloed JD, Hershberger RE, Morales A, Judge DP, Syrris P, Pilichou K, Domingo AM, Murray B, Cadrin-Tourigny J, et al. International evidence based reappraisal of genes associated with arrhythmogenic right ventricular cardiomyopathy using the clinical genome resource framework. Circ Genom Precis Med. 2021;14:e003273. doi: 10.1161/CIRCGEN.120.003273 - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
