

Defective Desmosomal Adhesion Causes Arrhythmogenic Cardiomyopathy by Involving an Integrin-αVβ6/TGF-β Signaling Cascade
- PMID: 36268721
- PMCID: PMC9674449
- DOI: 10.1161/CIRCULATIONAHA.121.057329
Defective Desmosomal Adhesion Causes Arrhythmogenic Cardiomyopathy by Involving an Integrin-αVβ6/TGF-β Signaling Cascade
Abstract
Background: Arrhythmogenic cardiomyopathy (ACM) is characterized by progressive loss of cardiomyocytes with fibrofatty tissue replacement, systolic dysfunction, and life-threatening arrhythmias. A substantial proportion of ACM is caused by mutations in genes of the desmosomal cell-cell adhesion complex, but the underlying mechanisms are not well understood. In the current study, we investigated the relevance of defective desmosomal adhesion for ACM development and progression.
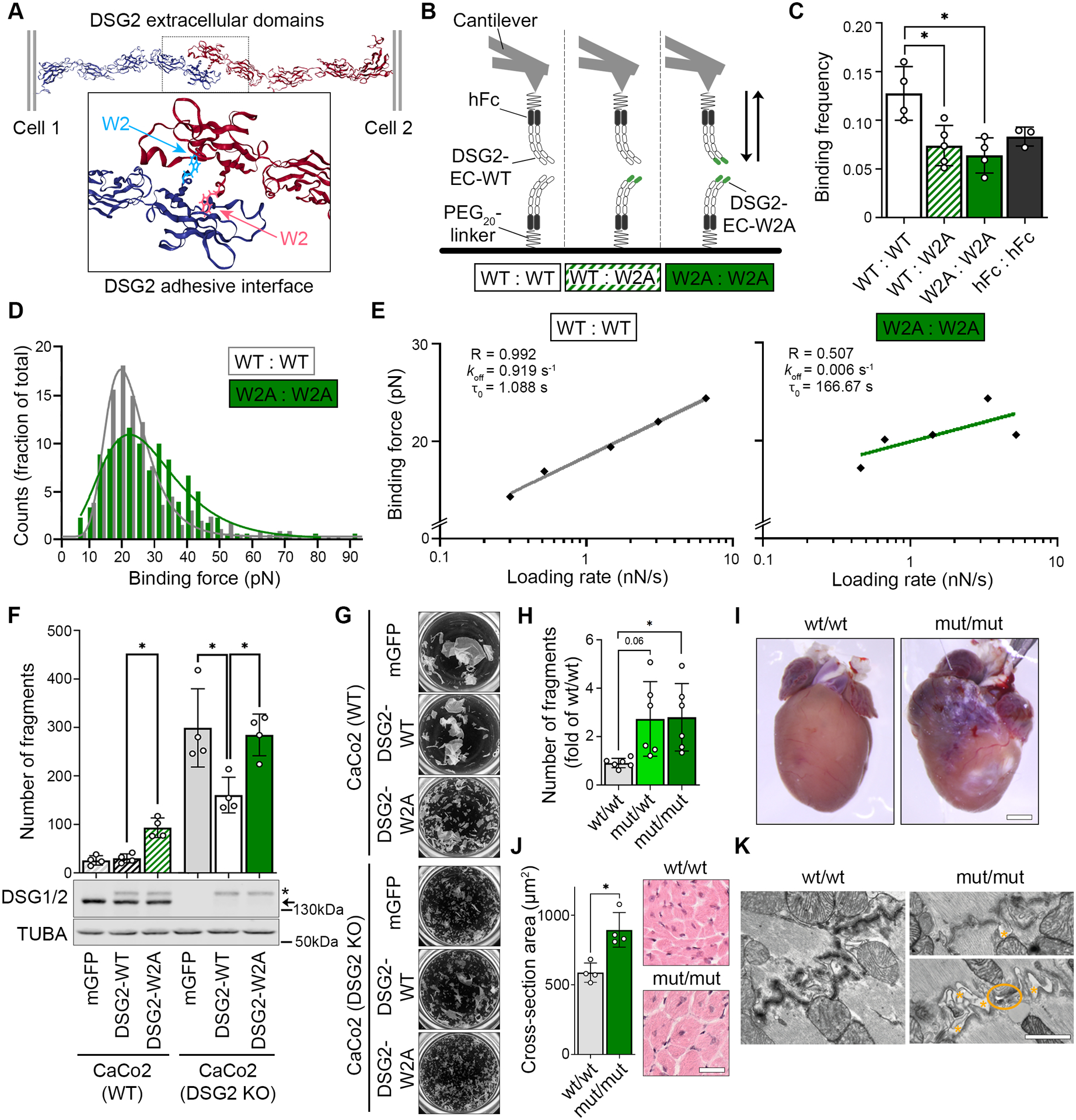
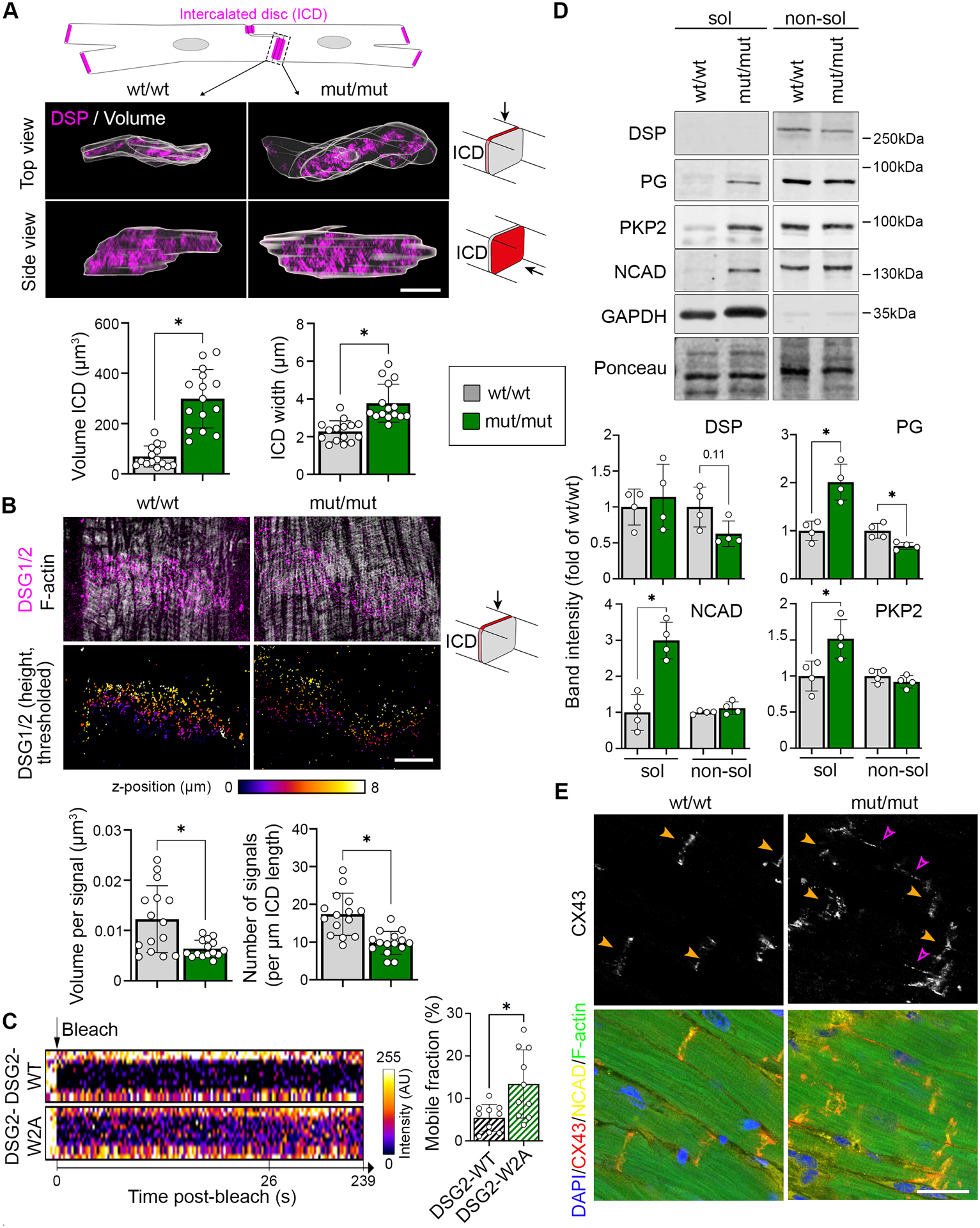
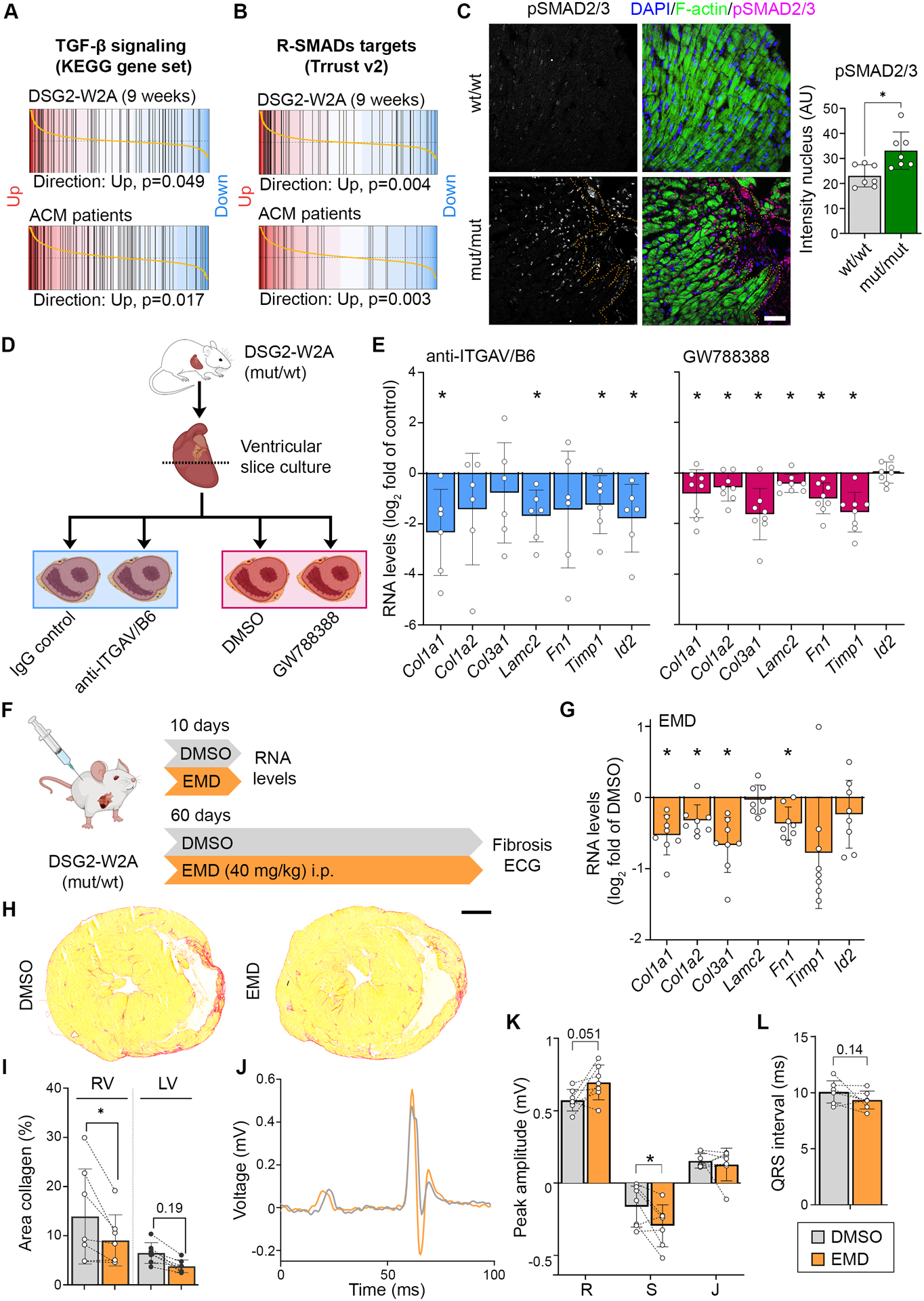
Methods: We mutated the binding site of DSG2 (desmoglein-2), a crucial desmosomal adhesion molecule in cardiomyocytes. This DSG2-W2A mutation abrogates the tryptophan swap, a central interaction mechanism of DSG2 on the basis of structural data. Impaired adhesive function of DSG2-W2A was confirmed by cell-cell dissociation assays and force spectroscopy measurements by atomic force microscopy. The DSG2-W2A knock-in mouse model was analyzed by echocardiography, ECG, and histologic and biomolecular techniques including RNA sequencing and transmission electron and superresolution microscopy. The results were compared with ACM patient samples, and their relevance was confirmed in vivo and in cardiac slice cultures by inhibitor studies applying the small molecule EMD527040 or an inhibitory integrin-αVβ6 antibody.
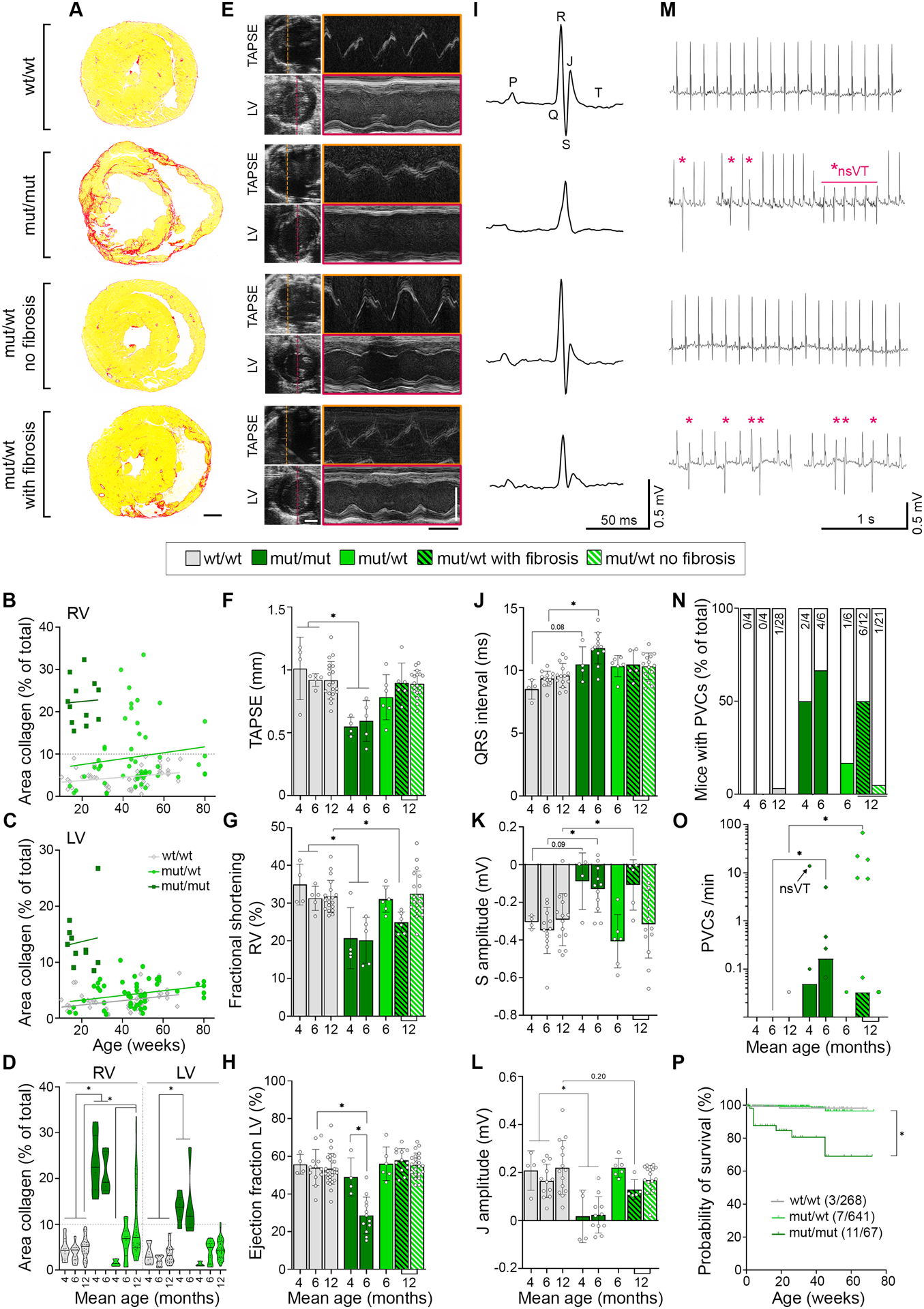
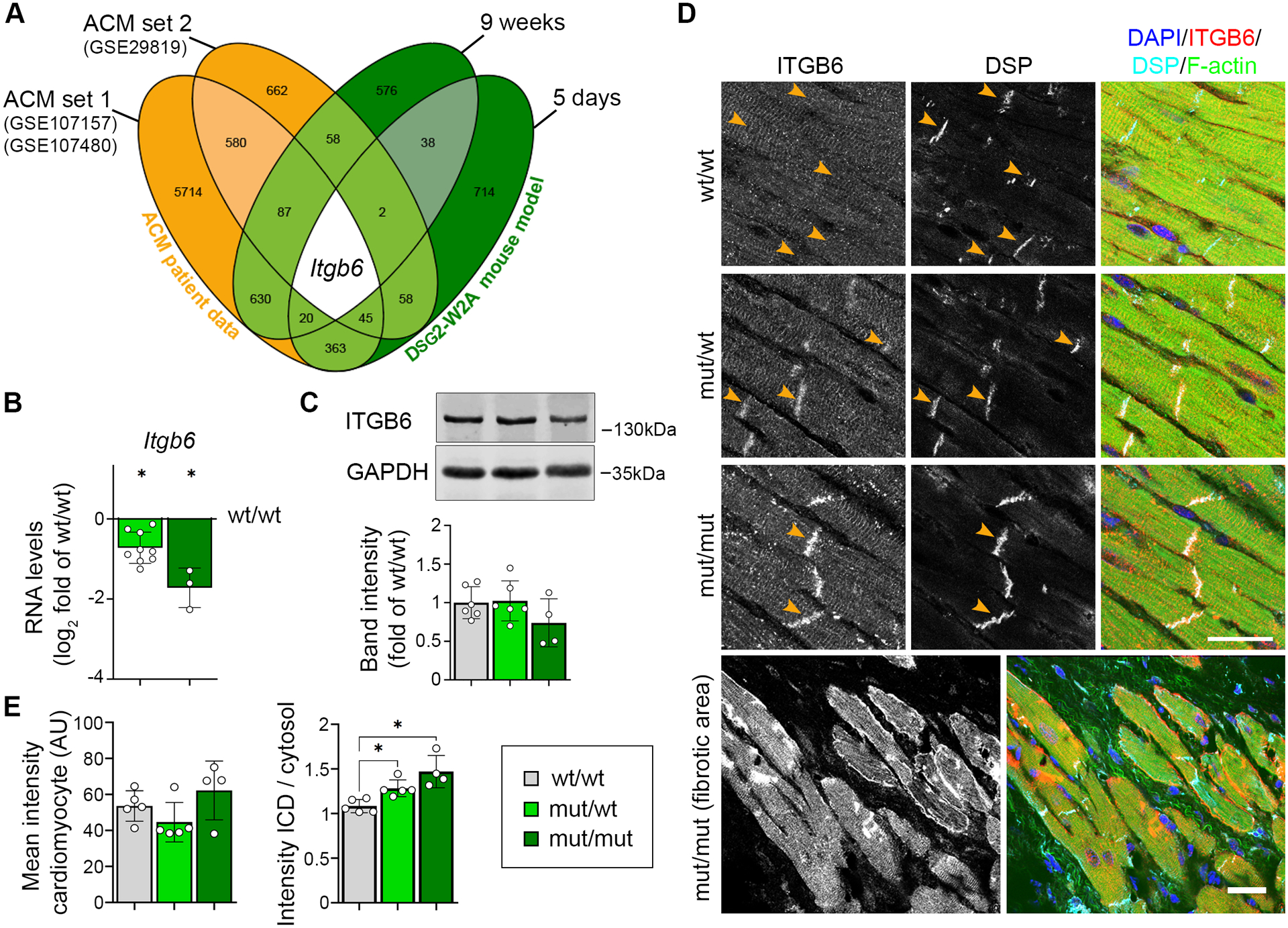
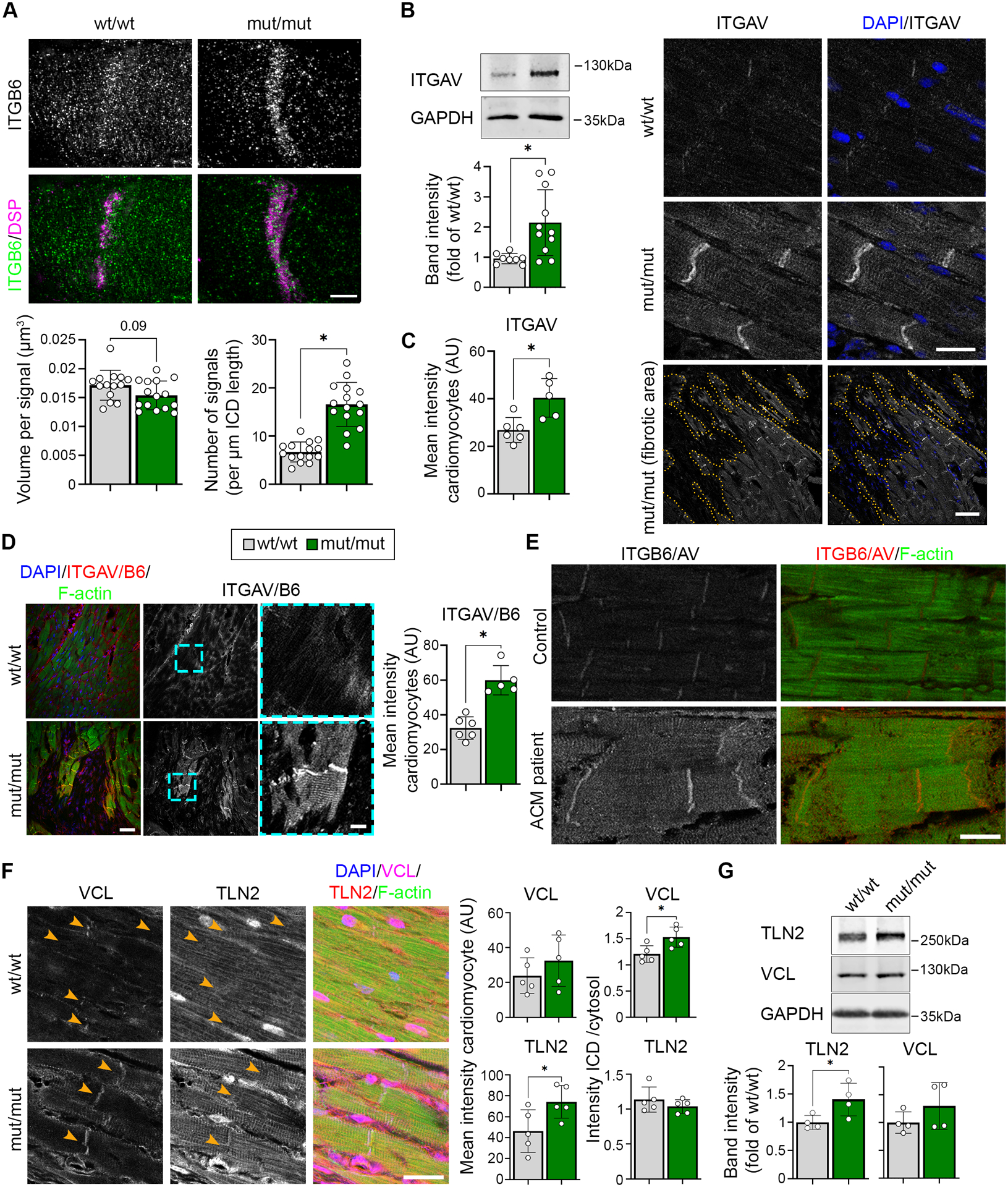
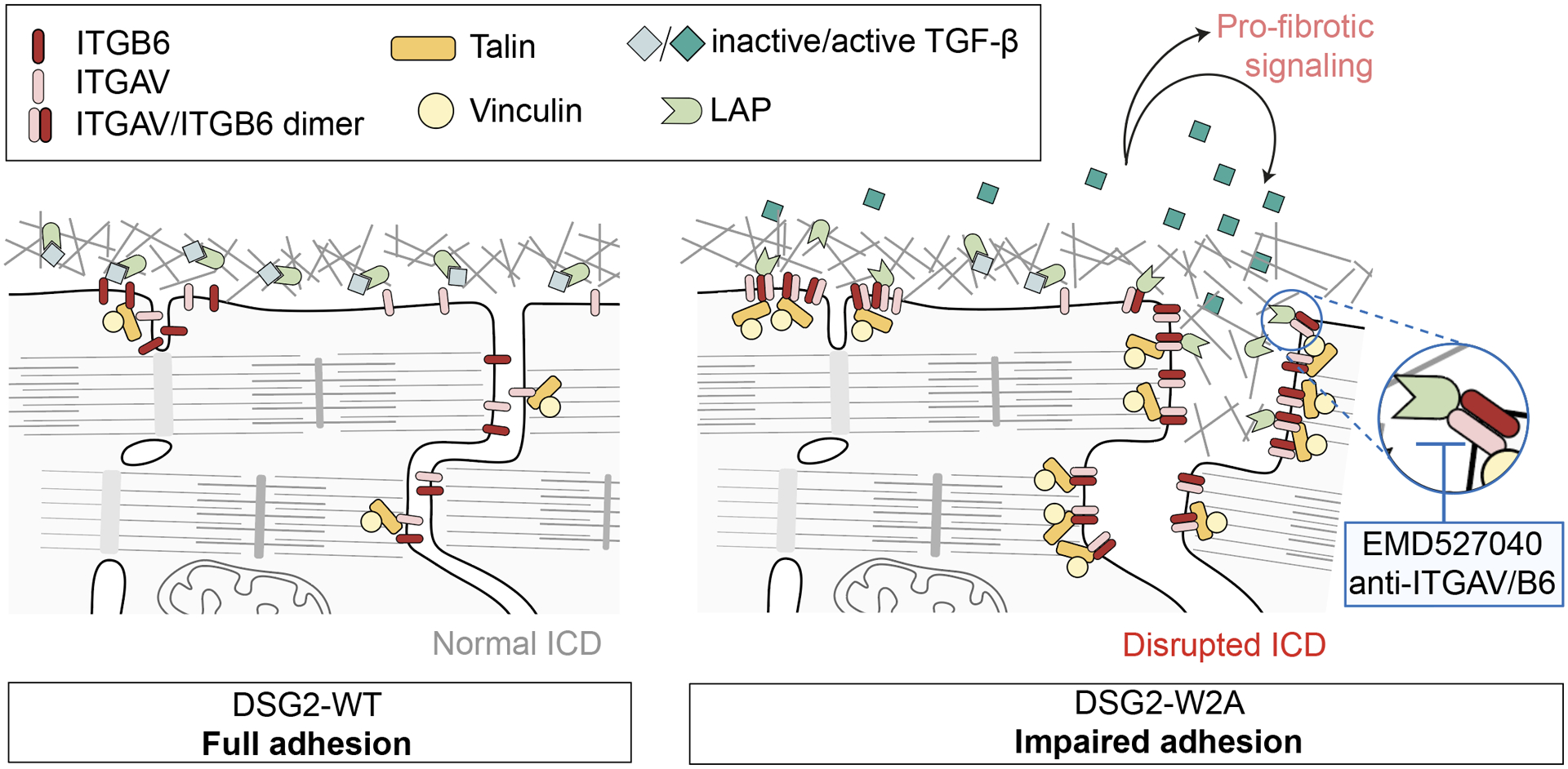
Results: The DSG2-W2A mutation impaired binding on molecular level and compromised intercellular adhesive function. Mice bearing this mutation develop a severe cardiac phenotype recalling the characteristics of ACM, including cardiac fibrosis, impaired systolic function, and arrhythmia. A comparison of the transcriptome of mutant mice with ACM patient data suggested deregulated integrin-αVβ6 and subsequent transforming growth factor-β signaling as driver of cardiac fibrosis. Blocking integrin-αVβ6 led to reduced expression of profibrotic markers and reduced fibrosis formation in mutant animals in vivo.
Conclusions: We show that disruption of desmosomal adhesion is sufficient to induce a phenotype that fulfils the clinical criteria to establish the diagnosis of ACM, confirming the dysfunctional adhesion hypothesis. Deregulation of integrin-αVβ6 and transforming growth factor-β signaling was identified as a central step toward fibrosis. A pilot in vivo drug test revealed this pathway as a promising target to ameliorate fibrosis. This highlights the value of this model to discern mechanisms of cardiac fibrosis and to identify and test novel treatment options for ACM.
Keywords: cardiomyopathies; desmosomes; fibrosis; integrins; intercalated disc; transforming growth factors.
Figures
References
-
- Basso C, Czarnowska E, Della Barbera M, Bauce B, Beffagna G, Wlodarska EK, Pilichou K, Ramondo A, Lorenzon A, Wozniek O, et al. Ultrastructural evidence of intercalated disc remodelling in arrhythmogenic right ventricular cardiomyopathy: an electron microscopy investigation on endomyocardial biopsies. Eur Heart J. 2006;27:1847–1854. doi: 10.1093/eurheartj/ehl095 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
