

Shared and distinct biological circuits in effector, memory and exhausted CD8+ T cells revealed by temporal single-cell transcriptomics and epigenetics
- PMID: 36271148
- PMCID: PMC10408358
- DOI: 10.1038/s41590-022-01338-4
Shared and distinct biological circuits in effector, memory and exhausted CD8+ T cells revealed by temporal single-cell transcriptomics and epigenetics
Abstract
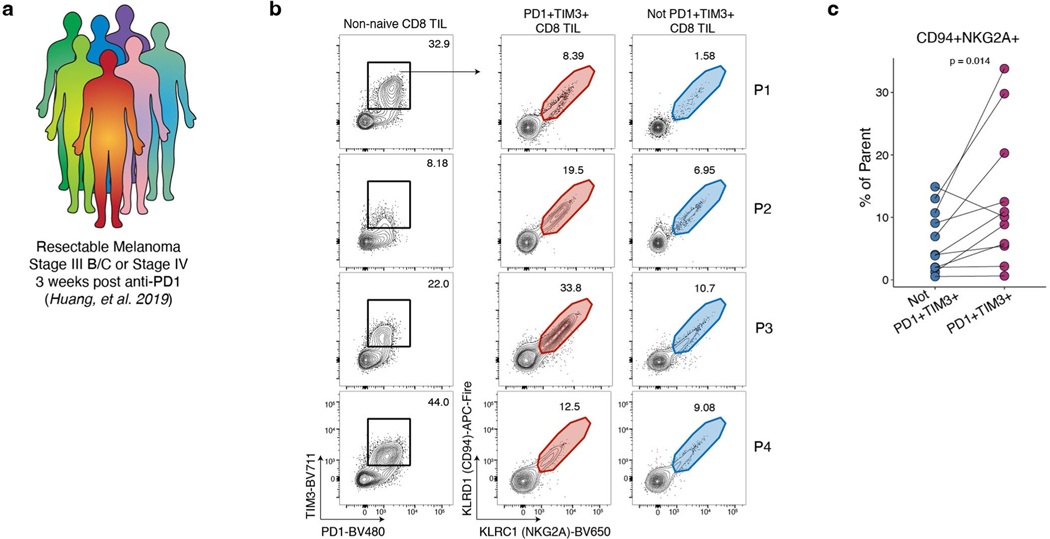
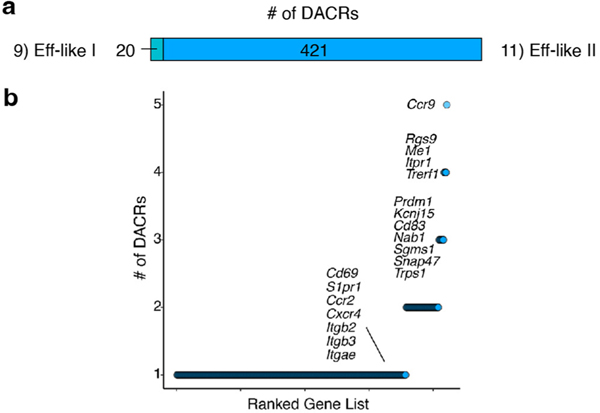
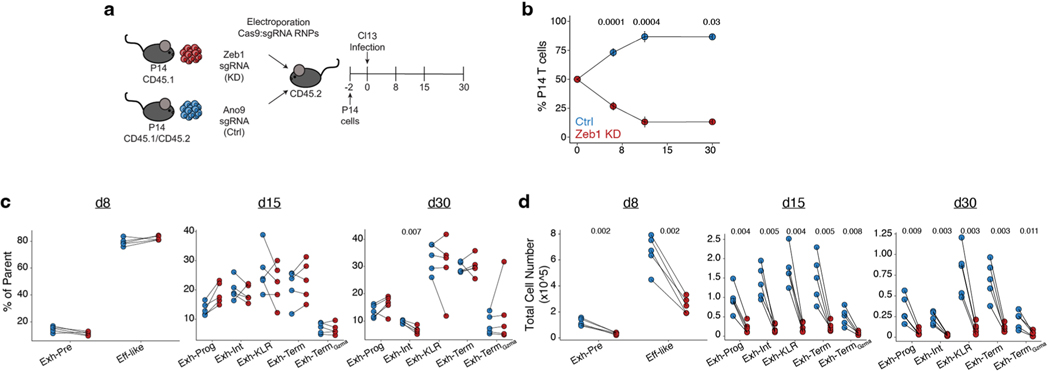
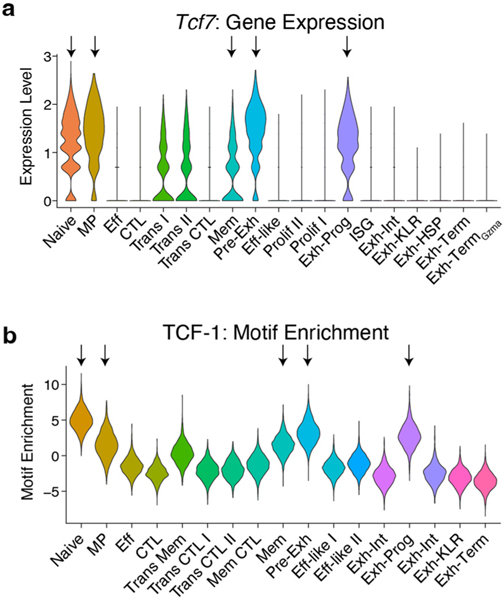
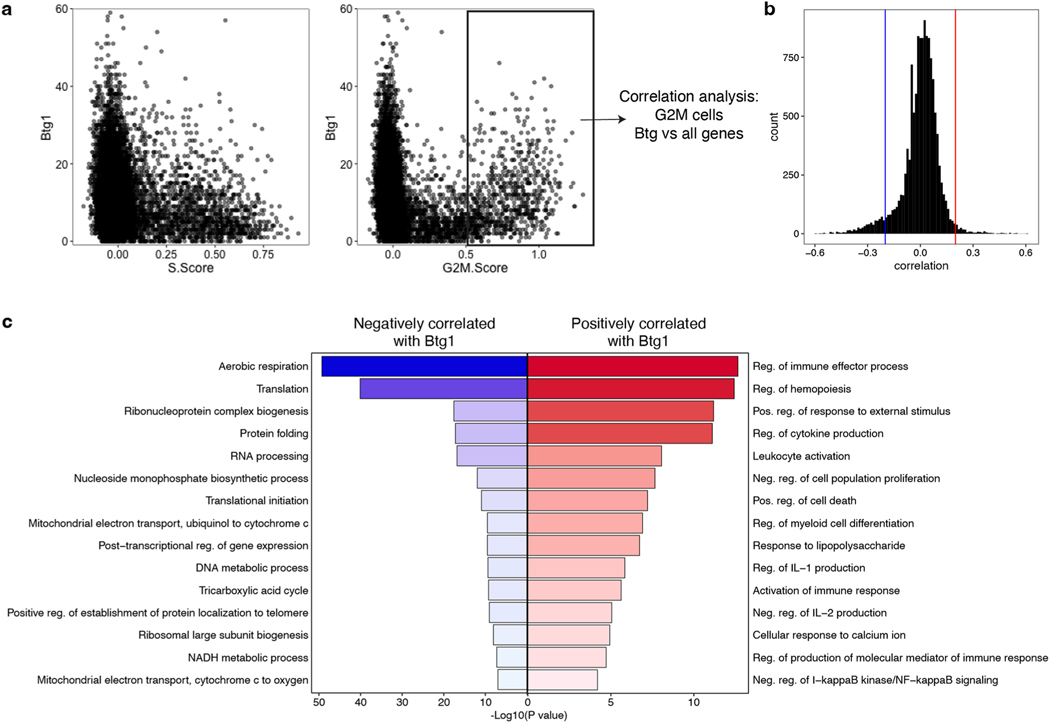
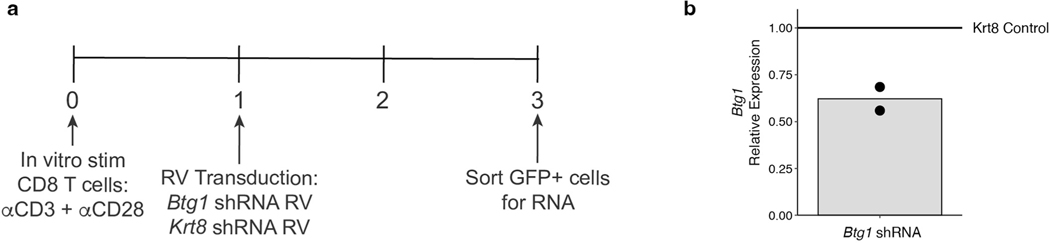
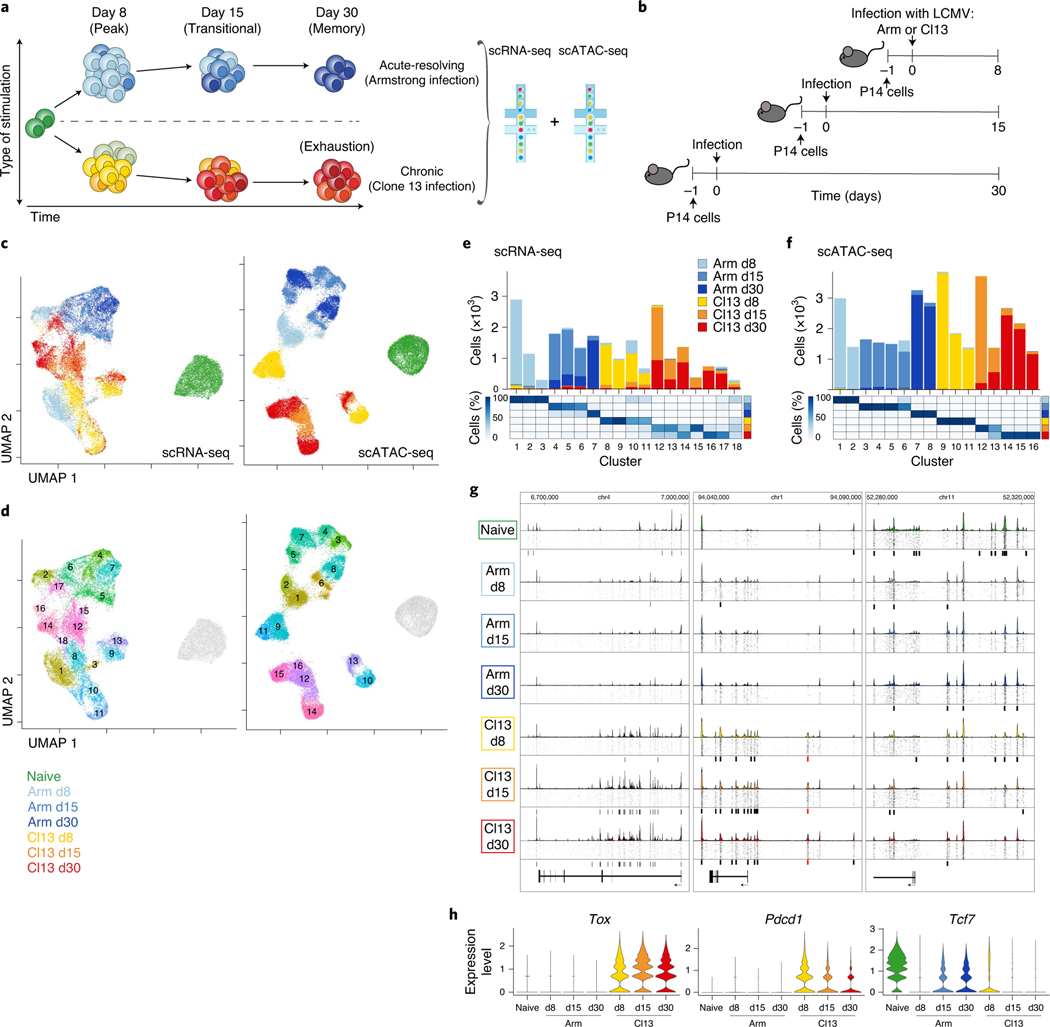
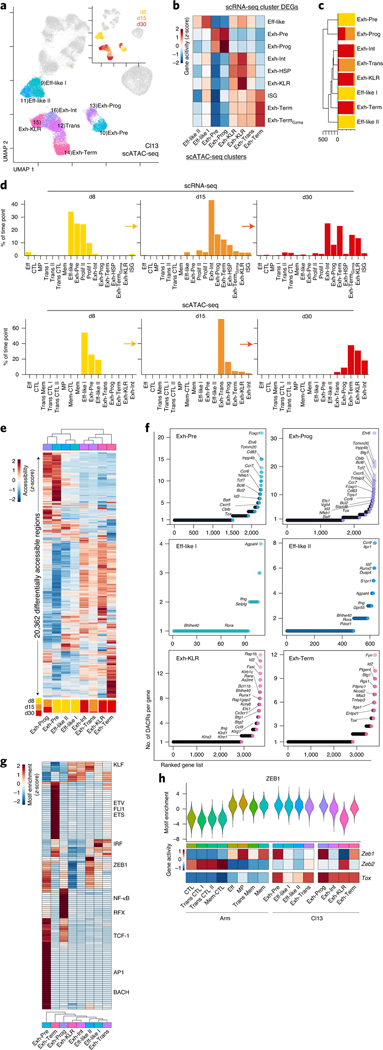
Naïve CD8+ T cells can differentiate into effector (Teff), memory (Tmem) or exhausted (Tex) T cells. These developmental pathways are associated with distinct transcriptional and epigenetic changes that endow cells with different functional capacities and therefore therapeutic potential. The molecular circuitry underlying these developmental trajectories and the extent of heterogeneity within Teff, Tmem and Tex populations remain poorly understood. Here, we used the lymphocytic choriomeningitis virus model of acute-resolving and chronic infection to address these gaps by applying longitudinal single-cell RNA-sequencing (scRNA-seq) and single-cell assay for transposase-accessible chromatin sequencing (scATAC-seq) analyses. These analyses uncovered new subsets, including a subpopulation of Tex cells expressing natural killer cell-associated genes that is dependent on the transcription factor Zeb2, as well as multiple distinct TCF-1+ stem/progenitor-like subsets in acute and chronic infection. These data also revealed insights into the reshaping of Tex subsets following programmed death 1 (PD-1) pathway blockade and identified a key role for the cell stress regulator, Btg1, in establishing the Tex population. Finally, these results highlighted how the same biological circuits such as cytotoxicity or stem/progenitor pathways can be used by CD8+ T cell subsets with highly divergent underlying chromatin landscapes generated during different infections.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
E.J.W. is a member of the Parker Institute for Cancer Immunotherapy, which supported the study. E.J.W. is an advisor for Danger Bio, Marengo, Janssen, Pluto Immunotherapeutics, Related Sciences, Rubius Therapeutics, Synthekine and Surface Oncology. E.J.W. is a founder of Surface Oncology, Danger Bio and Arsenal Biosciences. E.J.W. has a patent on the PD-1 pathway. O.K. holds equity in Arsenal Biosciences and is an employee of Orange Grove Bio. A.C.H. is a consultant for Immunai and receives funding from BMS. X.X. is scientific cofounder of CureBiotech and Exio Biosciences. T.M. is on the scientific advisory board for Merck, BMS, OncoSec, GigaGen and Instil Bio. G.C.K. is on the scientific advisory board for Merck and was the principal investigator of an investigator-initiated trial sponsored by Merck.
Figures
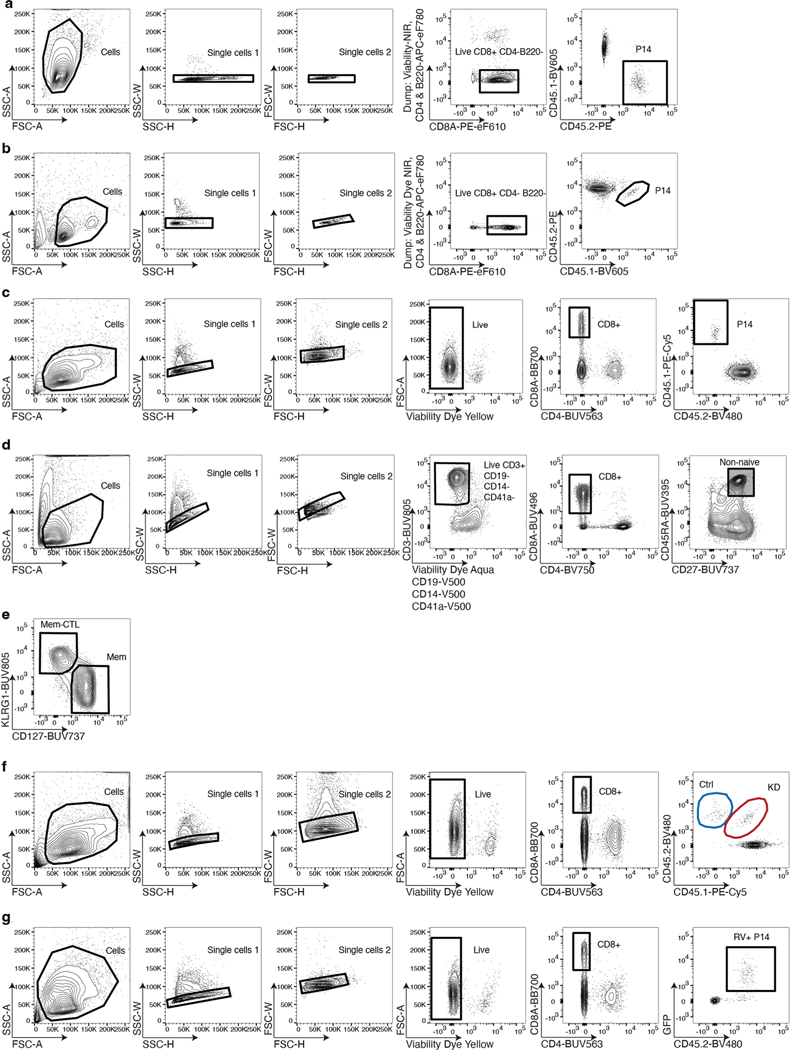
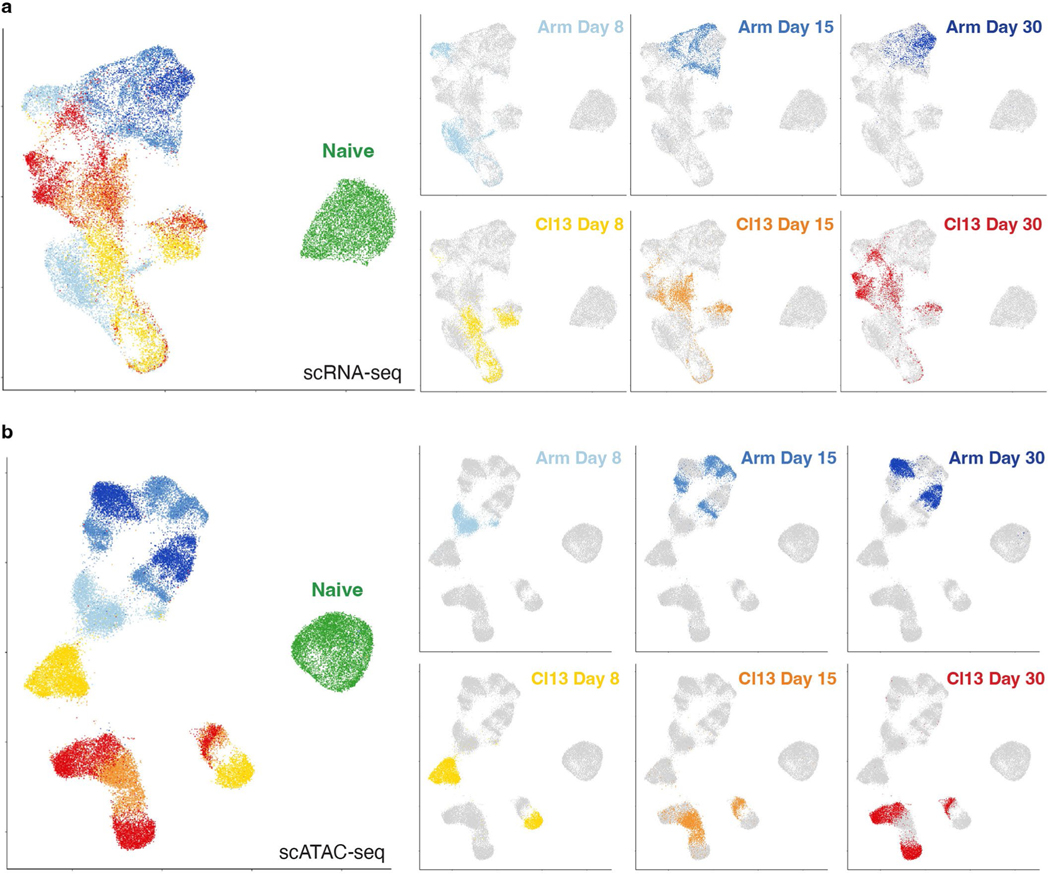
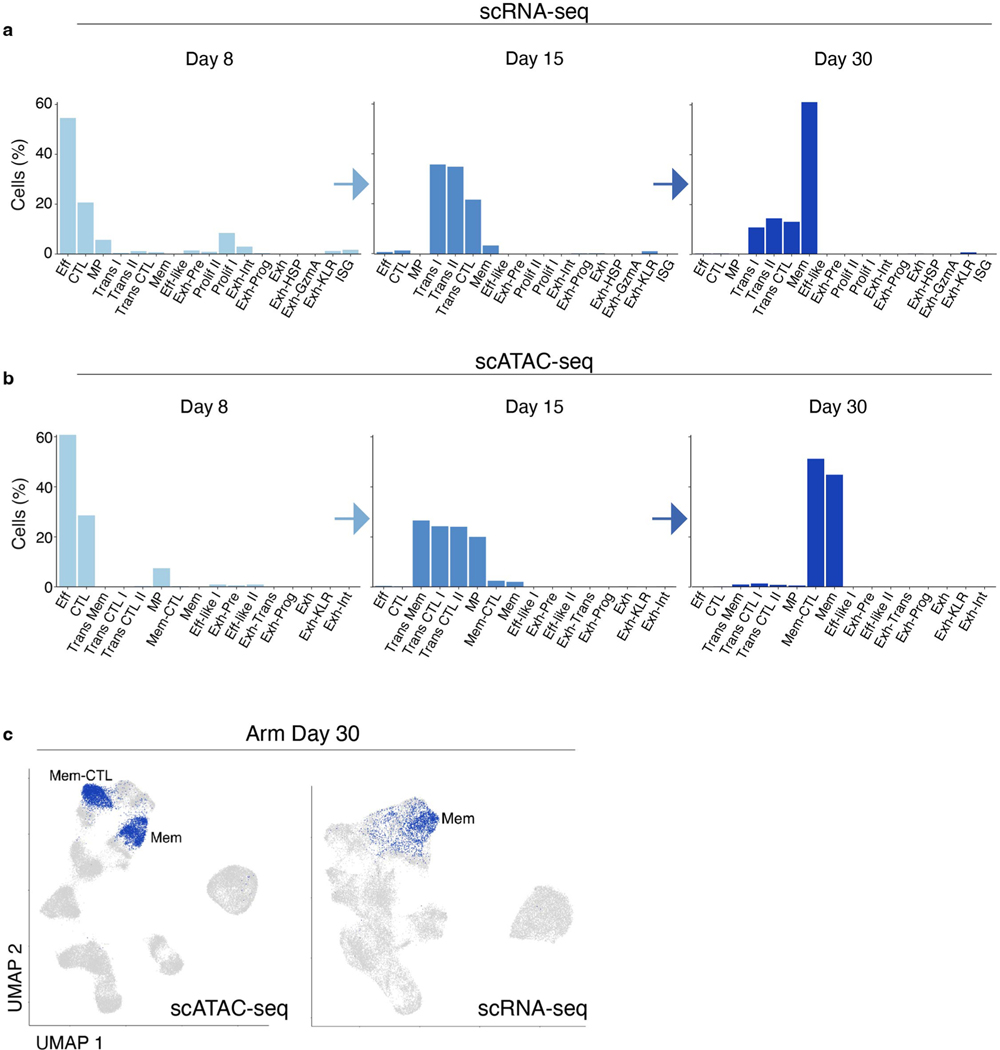
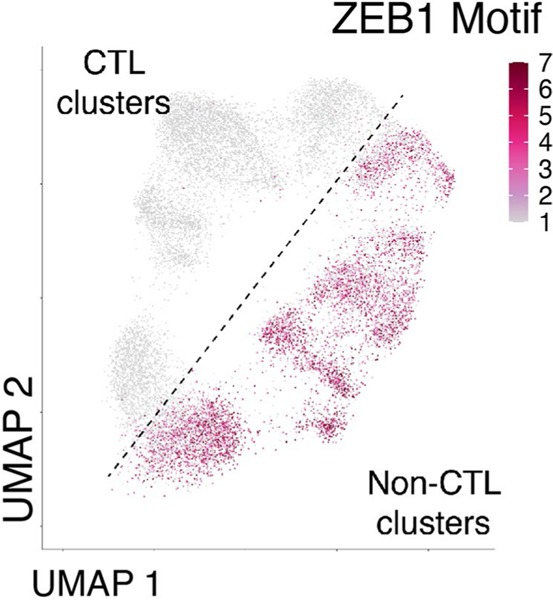


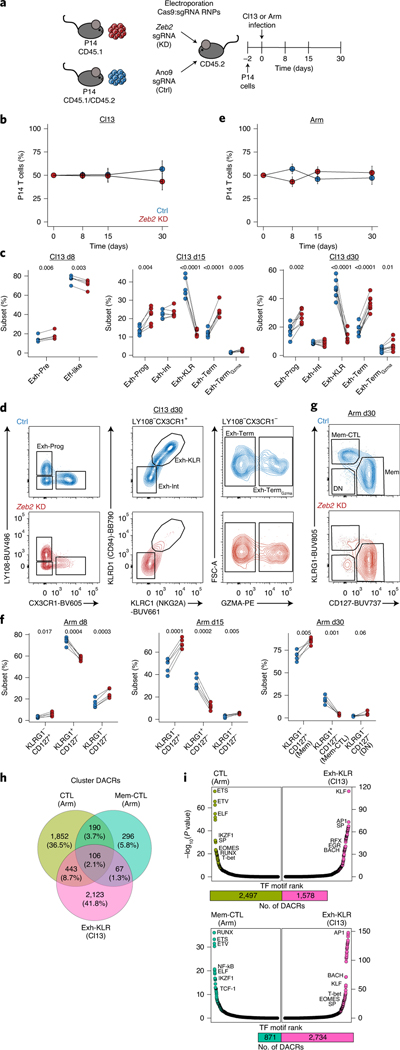
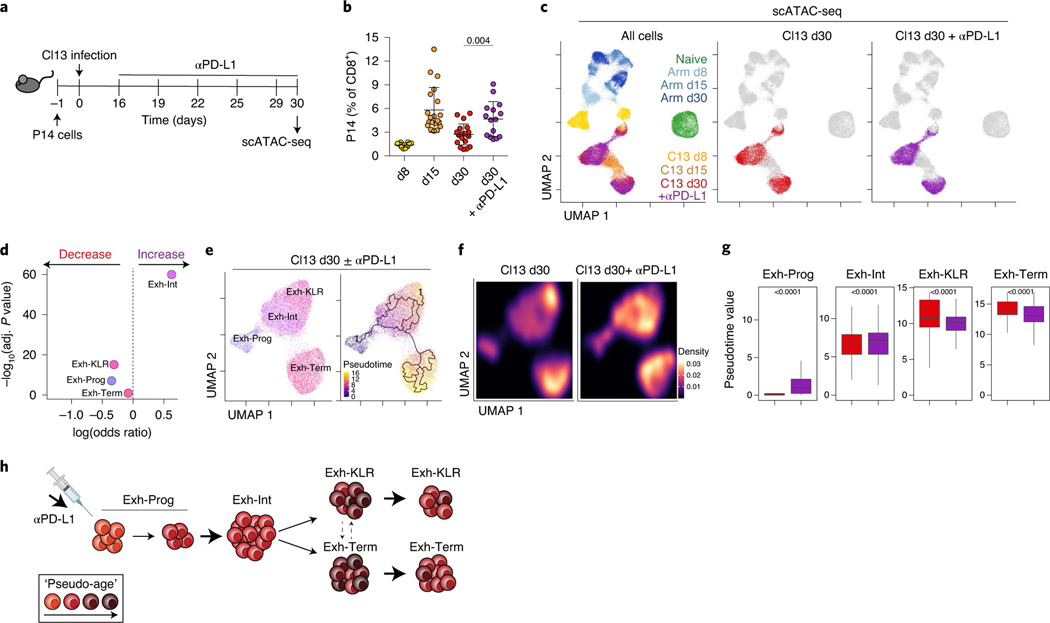
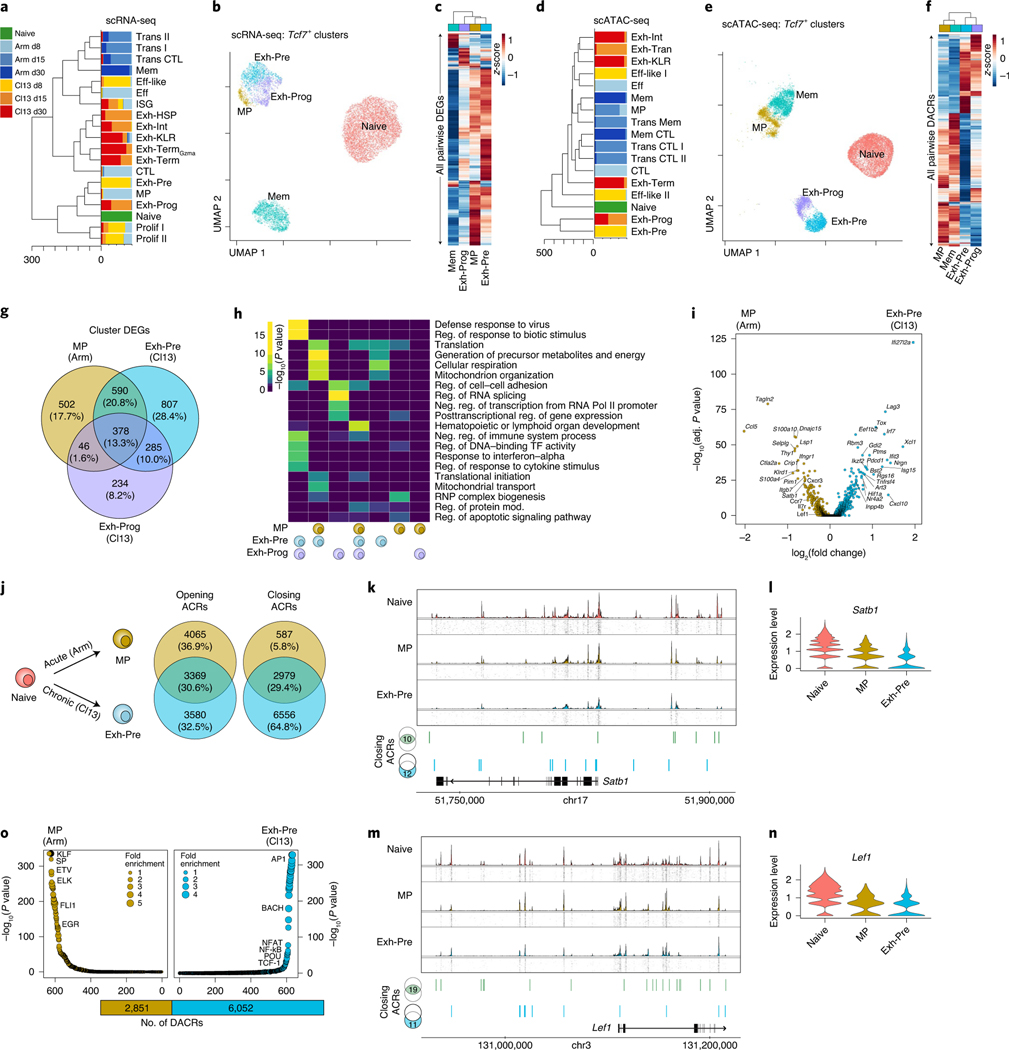
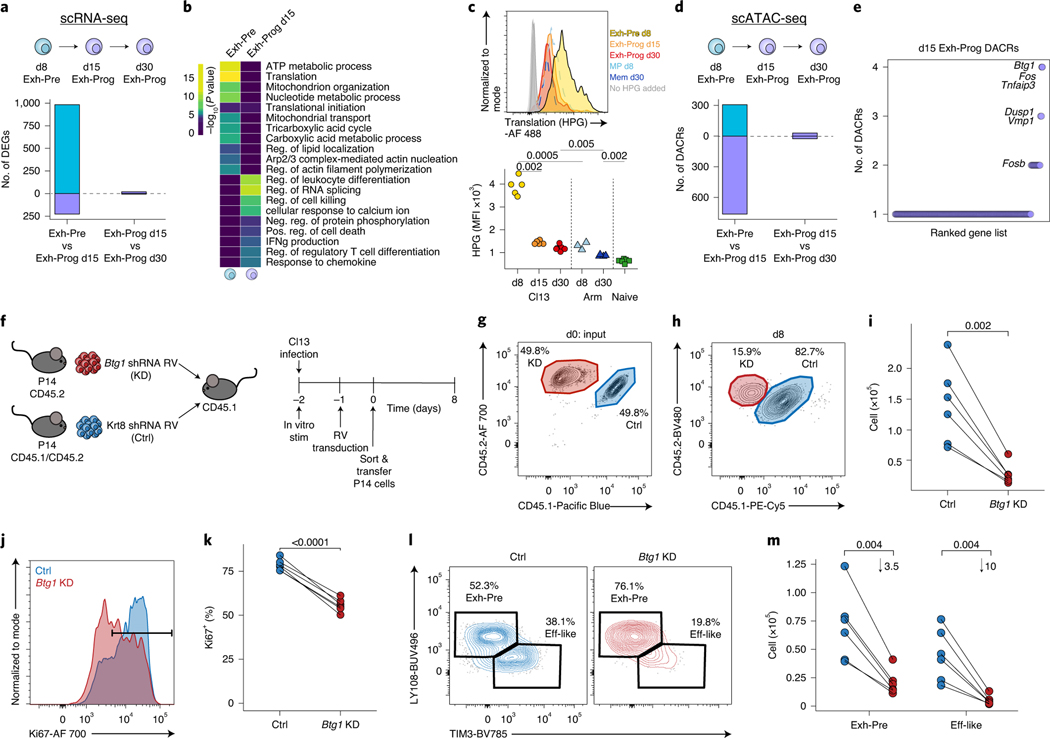
References
-
- Kaech SM et al. Selective expression of the interleukin-7 receptor identifies effector CD8+ T cells that give rise to long-lived memory cells. Nat. Immunol. 4, 1191–1198 (2003). - PubMed
-
- McLane LM, Abdel-Hakeem MS & Wherry EJ. CD8+ T cell exhaustion during chronic viral infection and cancer. Annu. Rev. Immunol. 37, 457–495 (2019). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P01 CA210944/CA/NCI NIH HHS/United States
- F30 AI129263/AI/NIAID NIH HHS/United States
- U01 DK127768/DK/NIDDK NIH HHS/United States
- U19 AI117950/AI/NIAID NIH HHS/United States
- K08 CA230157/CA/NCI NIH HHS/United States
- U19 AI082630/AI/NIAID NIH HHS/United States
- P50 CA174523/CA/NCI NIH HHS/United States
- P01 AI108545/AI/NIAID NIH HHS/United States
- T32 CA009140/CA/NCI NIH HHS/United States
- P50 CA261608/CA/NCI NIH HHS/United States
- R01 AI155577/AI/NIAID NIH HHS/United States
- U19 AI149680/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
