

Patients' view on gene therapy development for lysosomal storage disorders: a qualitative study
- PMID: 36271424
- PMCID: PMC9587648
- DOI: 10.1186/s13023-022-02543-y
Patients' view on gene therapy development for lysosomal storage disorders: a qualitative study
Abstract
Introduction: Several new treatment modalities are being developed for lysosomal storage disorders (LSDs), including gene therapy. As the currently available treatment options and their influence on disease progression differ greatly within the spectrum of LSDs, willingness to undergo gene therapy might vary among patients with LSDs and/or their representatives. The width of the LSD spectrum is illustrated by the differences between type 1 Gaucher disease, Fabry disease and Mucopolysaccharidosis type III (MPS III). For type 1 Gaucher and Fabry disease several therapies are available, resulting in a near normal or improved, but individually varying, prognosis. No treatment options are available for MPS III.
Aim: To identify factors influencing patients' and/or their representatives' decisions regarding undergoing gene therapy.
Methods: Focus group discussions and semi-structured interviews were conducted with patients with type 1 Gaucher disease, Fabry disease and MPS III. Parents of MPS III patients were included as patients' representatives.
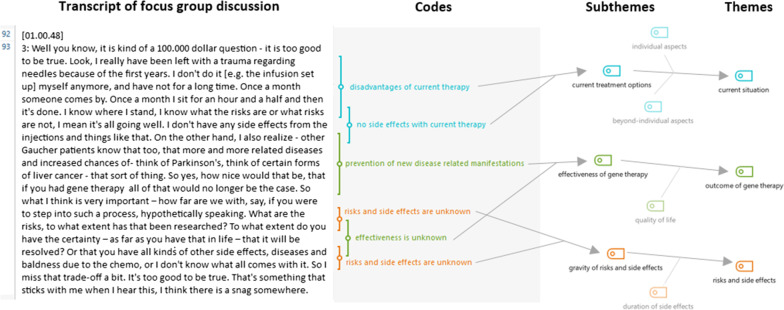
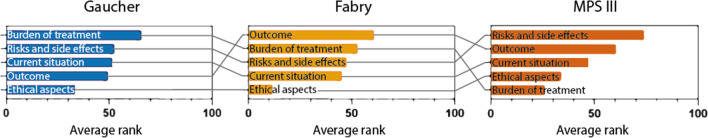
Results: Nine Gaucher patients, 23 Fabry patients, two adult MPS III patients and five parents of MPS III patients participated in the study. The five main themes that arose were: outcome of gene therapy, risks and side effects, burden of gene therapy treatment, current situation and ethical aspects. Participants' views ranged from hesitance to eagerness to undergo gene therapy, which seemed to be mostly related to disease severity and currently available treatment options. Severe disease, limited treatment options and limited effectiveness of current treatment augmented the willingness to choose gene therapy. Gaucher and Fabry patients deemed the burden of treatment important. Fabry and MPS III patients and parents considered outcome important, suggesting hope for improvement. When asked to rank the factors discussed in the focus group discussions, Gaucher patients ranked outcome low, which could indicate a more cautious attitude towards gene therapy.
Conclusion: This study underlines the importance of exploring patients' needs and expectations before using limited resources in the development of therapies for patient groups of which a significant subset may not be willing to undergo that specific therapy.
Keywords: Fabry disease; Focus group discussions; Gaucher disease type 1; Gene therapy; Lysosomal storage diseases; Mucopolysaccharidosis type III; Qualitative research.
© 2022. The Author(s).
Conflict of interest statement
CB, EC, TM, HD, and EM have nothing to disclose. EE is involved as a sub-investigator in a pre-marketing study with Sanofi Genzyme. MB was a sub-investigator in the Lysogene gene therapy study for MPS III (NCT02053064). ML and CH are involved in premarketing studies with Sanofi-Genzyme, Protalix and Idorsia. BS is involved in a premarketing study with Protalix.
Figures
Similar articles
-
Different diseases, different needs: Patient preferences for gene therapy in lysosomal storage disorders, a probabilistic threshold technique survey.Orphanet J Rare Dis. 2024 Oct 3;19(1):367. doi: 10.1186/s13023-024-03371-y. Orphanet J Rare Dis. 2024. PMID: 39363355 Free PMC article.
-
Treating lysosomal storage disorders: What have we learnt?J Inherit Metab Dis. 2020 Jan;43(1):125-132. doi: 10.1002/jimd.12131. Epub 2019 Jun 26. J Inherit Metab Dis. 2020. PMID: 31140601 Review.
-
A charitable access program for patients with lysosomal storage disorders in underserved communities worldwide.Orphanet J Rare Dis. 2021 Jan 6;16(1):8. doi: 10.1186/s13023-020-01645-9. Orphanet J Rare Dis. 2021. PMID: 33407729 Free PMC article.
-
Pulmonary involvement in selected lysosomal storage diseases and the impact of enzyme replacement therapy: A state-of-the art review.Clin Respir J. 2020 May;14(5):422-429. doi: 10.1111/crj.13150. Epub 2020 Jan 22. Clin Respir J. 2020. PMID: 31912638 Review.
-
Treatable lysosomal storage diseases in the advent of disease-specific therapy.Intern Med J. 2020 Nov;50 Suppl 4:5-27. doi: 10.1111/imj.15100. Intern Med J. 2020. PMID: 33210402
Cited by
-
Different diseases, different needs: Patient preferences for gene therapy in lysosomal storage disorders, a probabilistic threshold technique survey.Orphanet J Rare Dis. 2024 Oct 3;19(1):367. doi: 10.1186/s13023-024-03371-y. Orphanet J Rare Dis. 2024. PMID: 39363355 Free PMC article.
-
Treatment Beliefs Reflect Unmet Clinical Needs in Lysosomal Storage Diseases: An Opportunity for a Patient-Centered Approach.JIMD Rep. 2025 Feb 26;66(2):e70003. doi: 10.1002/jmd2.70003. eCollection 2025 Mar. JIMD Rep. 2025. PMID: 40017528 Free PMC article.
References
-
- Hollak CE, Lachmann R. Inherited metabolic disease in adults: a clinical guide. Oxford: Oxford University Press; 2016.
-
- Zimran A, Elstein D. Management of Gaucher disease: enzyme replacement therapy. Pediatr Endocrinol Rev. 2014;12(Suppl 1):82–87. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
