

SPiP: Splicing Prediction Pipeline, a machine learning tool for massive detection of exonic and intronic variant effects on mRNA splicing
- PMID: 36273432
- PMCID: PMC10946553
- DOI: 10.1002/humu.24491
SPiP: Splicing Prediction Pipeline, a machine learning tool for massive detection of exonic and intronic variant effects on mRNA splicing
Abstract
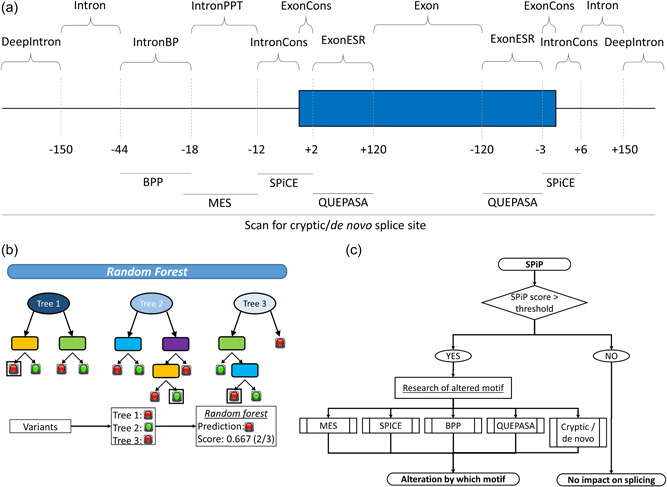
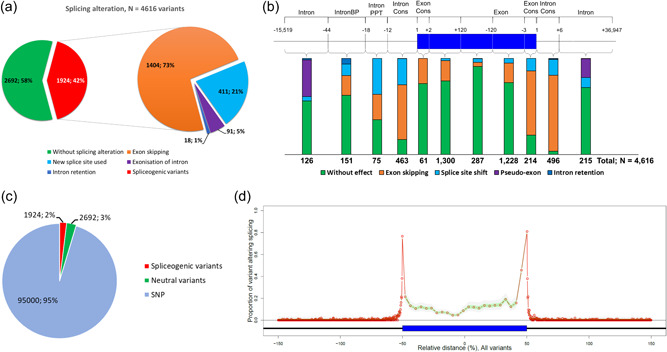
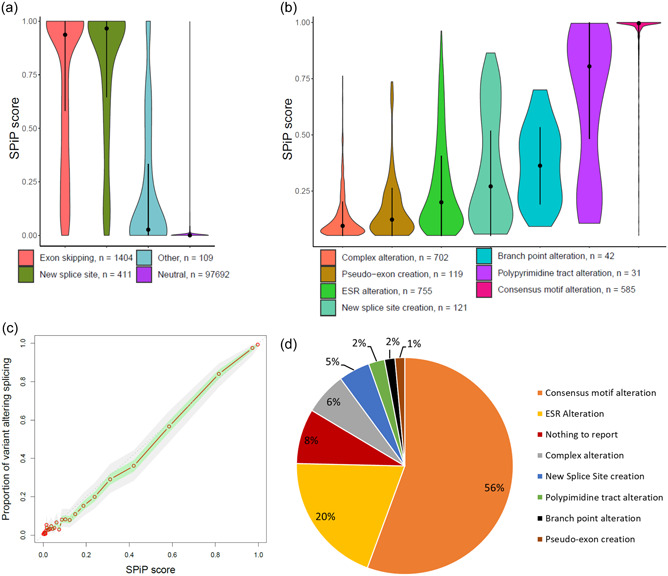
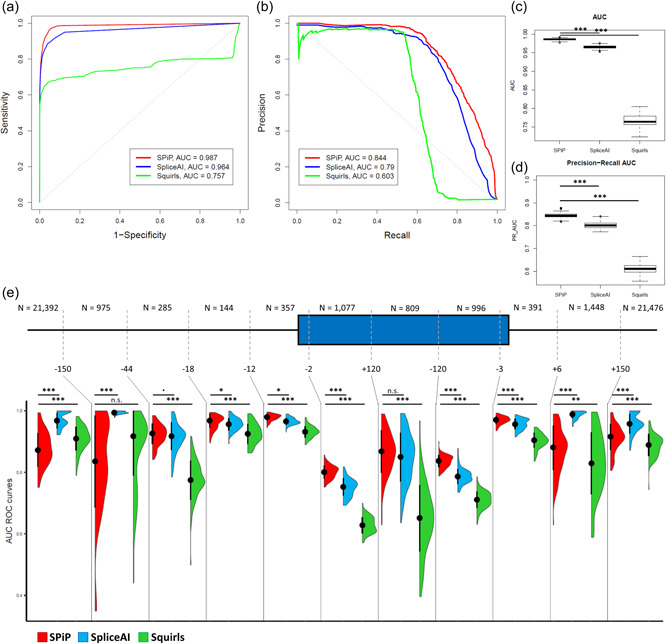
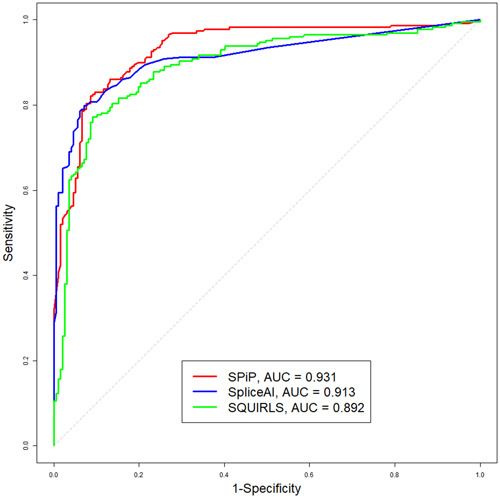
Modeling splicing is essential for tackling the challenge of variant interpretation as each nucleotide variation can be pathogenic by affecting pre-mRNA splicing via disruption/creation of splicing motifs such as 5'/3' splice sites, branch sites, or splicing regulatory elements. Unfortunately, most in silico tools focus on a specific type of splicing motif, which is why we developed the Splicing Prediction Pipeline (SPiP) to perform, in one single bioinformatic analysis based on a machine learning approach, a comprehensive assessment of the variant effect on different splicing motifs. We gathered a curated set of 4616 variants scattered all along the sequence of 227 genes, with their corresponding splicing studies. The Bayesian analysis provided us with the number of control variants, that is, variants without impact on splicing, to mimic the deluge of variants from high-throughput sequencing data. Results show that SPiP can deal with the diversity of splicing alterations, with 83.13% sensitivity and 99% specificity to detect spliceogenic variants. Overall performance as measured by area under the receiving operator curve was 0.986, better than SpliceAI and SQUIRLS (0.965 and 0.766) for the same data set. SPiP lends itself to a unique suite for comprehensive prediction of spliceogenicity in the genomic medicine era. SPiP is available at: https://sourceforge.net/projects/splicing-prediction-pipeline/.
Keywords: RNA; SPiP; machine learning; sequence variants; splicing predictions.
© 2022 The Authors. Human Mutation published by Wiley Periodicals LLC.
Conflict of interest statement
H. T. was employed by Interactive Biosoftware for the time period October 2015–September 2018 in the context of a public–private PhD project (CIFRE fellowship #2015/0335) partnership between INSERM and Interactive Biosoftware. The remaining authors declare no conflict of interest.
Figures
References
-
- Buratti, E. , Chivers, M. , Královičová, J. , Romano, M. , Baralle, M. , Krainer, A. R. , & Vořechovský, I. (2007). Aberrant 5′ splice sites in human disease genes: Mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization. Nucleic Acids Research, 35, 4250–4263. - PMC - PubMed
-
- Callebaut, I. , Joubrel, R. , Pissard, S. , Kannengiesser, C. , Gerolami, V. , Ged, C. , Cadet, E. , Cartault, F. , Ka, C. , Gourlaouen, I. , Gourhant, L. , Oudin, C. , Goossens, M. , Grandchamp, B. , De Verneuil, H. , Rochette, J. , Ferec, C. , & Le Gac, G. (2014). Comprehensive functional annotation of 18 missense mutations found in suspected hemochromatosis type 4 patients. Human Molecular Genetics, 23, 4479–4490. - PubMed
-
- Casadei, S. , Gulsuner, S. , Shirts, B. H. , Mandell, J. B. , Kortbawi, H. M. , Norquist, B. S. , Swisher, E. M. , Lee, M. K. , Goldberg, Y. , O'Connor, R. , Tan, Z. , Pritchard, C. C. , King, M. C. , & Walsh, T. (2019). Characterization of splice‐altering mutations in inherited predisposition to cancer. Proceedings of the National Academy of Sciences, 116, 26798–26807. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
