

Smooth muscle Acid-sensing ion channel 1a as a therapeutic target to reverse hypoxic pulmonary hypertension
- PMID: 36275633
- PMCID: PMC9581175
- DOI: 10.3389/fmolb.2022.989809
Smooth muscle Acid-sensing ion channel 1a as a therapeutic target to reverse hypoxic pulmonary hypertension
Abstract
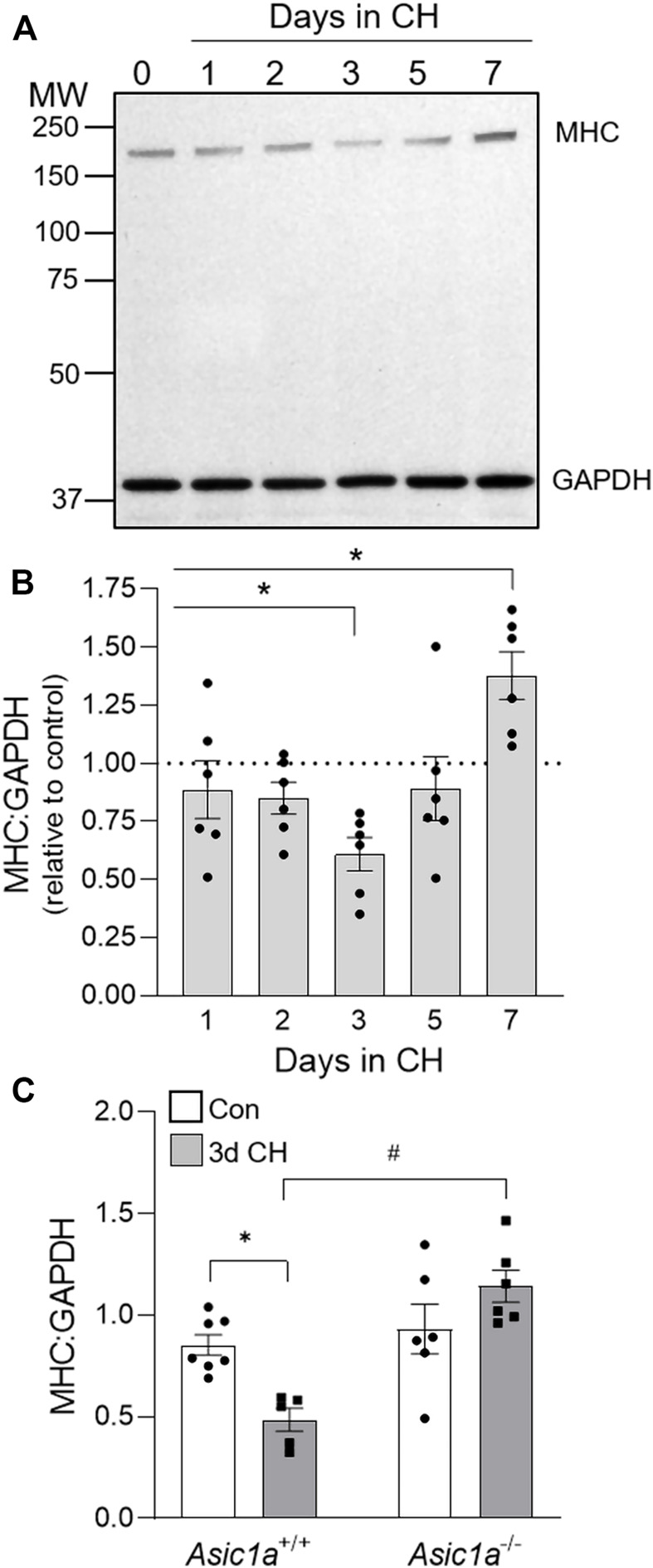
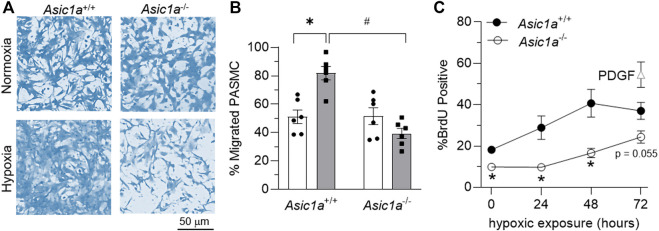
Acid-sensing ion channel 1a (ASIC1a) is a voltage-independent, non-selective cation channel that conducts both Na+ and Ca2+. Activation of ASIC1a elicits plasma membrane depolarization and stimulates intracellular Ca2+-dependent signaling pathways in multiple cell types, including vascular smooth muscle (SM) and endothelial cells (ECs). Previous studies have shown that increases in pulmonary vascular resistance accompanying chronic hypoxia (CH)-induced pulmonary hypertension requires ASIC1a to elicit enhanced pulmonary vasoconstriction and vascular remodeling. Both SM and EC dysfunction drive these processes; however, the involvement of ASIC1a within these different cell types is unknown. Using the Cre-LoxP system to generate cell-type-specific Asic1a knockout mice, we tested the hypothesis that SM-Asic1a contributes to CH-induced pulmonary hypertension and vascular remodeling, whereas EC-Asic1a opposes the development of CH-induced pulmonary hypertension. The severity of pulmonary hypertension was not altered in mice with specific deletion of EC-Asic1a (TekCre-Asic1a fl/fl). However, similar to global Asic1a knockout (Asic1a -/-) mice, mice with specific deletion of SM-Asic1a (MHCCreER-Asic1a fl/fl) were protected from the development of CH-induced pulmonary hypertension and right heart hypertrophy. Furthermore, pulmonary hypertension was reversed when deletion of SM-Asic1a was initiated in conditional MHCCreER-Asic1a fl/fl mice with established pulmonary hypertension. CH-induced vascular remodeling was also significantly attenuated in pulmonary arteries from MHCCreER-Asic1a fl/fl mice. These findings were additionally supported by decreased CH-induced proliferation and migration of pulmonary arterial smooth muscle cells (PASMCs) from Asic1a -/- mice. Together these data demonstrate that SM-, but not EC-Asic1a contributes to CH-induced pulmonary hypertension and vascular remodeling. Furthermore, these studies provide evidence for the therapeutic potential of ASIC1a inhibition to reverse pulmonary hypertension.
Keywords: endothelium; migration; proliferation; right heart hypertrophy; vascular remodeling.
Copyright © 2022 Garcia, Yellowhair, Detweiler, Ahmadian, Herbert, Gonzalez Bosc, Resta and Jernigan.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures






References
-
- Bierer R., Nitta C. H., Friedman J., Codianni S., de Frutos S., Dominguez-Bautista J. A., et al. (2011). NFATc3 is required for chronic hypoxia-induced pulmonary hypertension in adult and neonatal mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 301 (6), L872–L880. 10.1152/ajplung.00405.2010 - DOI - PMC - PubMed
-
- Bonnet S., Michelakis E. D., Porter C. J., Andrade-Navarro M. A., Thébaud B., Bonnet S., et al. (2006). An abnormal mitochondrial-hypoxia inducible factor-1alpha-kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: Similarities to human pulmonary arterial hypertension. Circulation 113 (22), 2630–2641. 10.1161/circulationaha.105.609008 - DOI - PubMed
-
- Broughton B. R., Jernigan N. L., Norton C. E., Walker B. R., Resta T. C. (2010). Chronic hypoxia augments depolarization-induced Ca2+ sensitization in pulmonary vascular smooth muscle through superoxide-dependent stimulation of RhoA. Am. J. Physiol. Lung Cell. Mol. Physiol. 298 (2), L232–L242. 10.1152/ajplung.00276.2009 - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous