

Large 22q13.3 deletions perturb peripheral transcriptomic and metabolomic profiles in Phelan-McDermid syndrome
- PMID: 36276299
- PMCID: PMC9579712
- DOI: 10.1016/j.xhgg.2022.100145
Large 22q13.3 deletions perturb peripheral transcriptomic and metabolomic profiles in Phelan-McDermid syndrome
Abstract
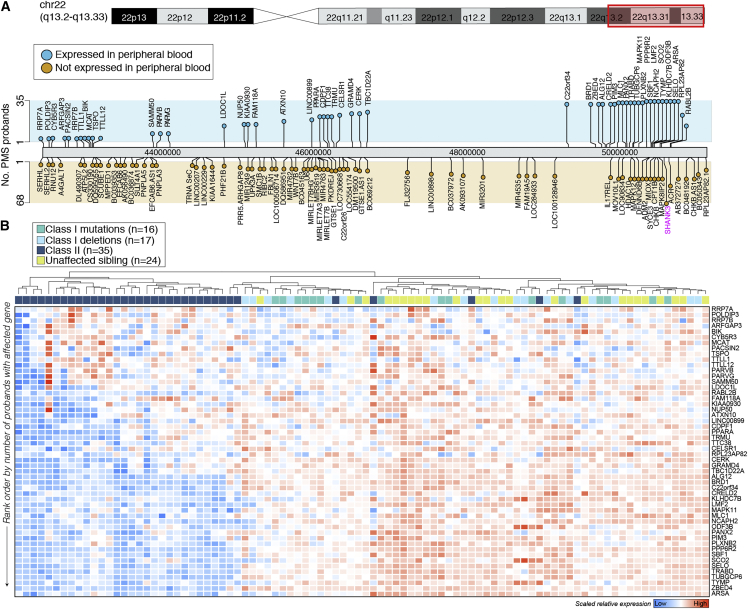
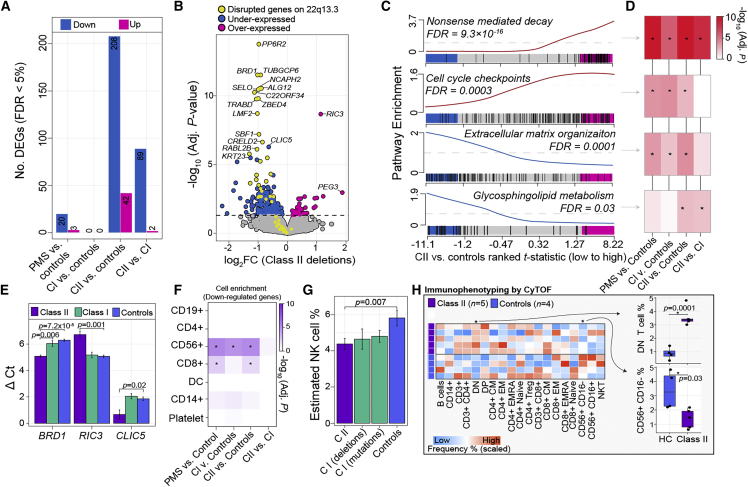
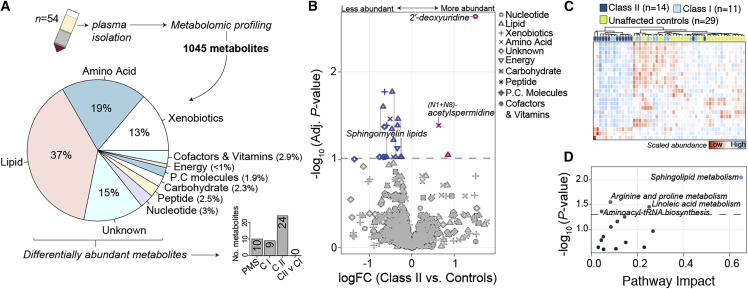
Phelan-McDermid syndrome (PMS) is a rare neurodevelopmental disorder caused at least in part by haploinsufficiency of the SHANK3 gene, due to sequence variants in SHANK3 or subtelomeric 22q13.3 deletions. Phenotypic differences have been reported between PMS participants carrying small "class I" mutations and large "class II" mutations; however, the molecular perturbations underlying these divergent phenotypes remain obscure. Using peripheral blood transcriptome and serum metabolome profiling, we examined the molecular perturbations in the peripheral circulation associated with a full spectrum of PMS genotypes spanning class I (n = 37) and class II mutations (n = 39). Transcriptomic data revealed 52 genes with blood expression profiles that tightly scale with 22q.13.3 deletion size. Furthermore, we uncover 208 underexpressed genes in PMS participants with class II mutations, which were unchanged in class I mutations. These genes were not linked to 22q13.3 and were strongly enriched for glycosphingolipid metabolism, NCAM1 interactions, and cytotoxic natural killer (NK) immune cell signatures. In silico predictions estimated a reduction in CD56+ CD16- NK cell proportions in class II mutations, which was validated by mass cytometry time of flight. Global metabolomics profiling identified 24 metabolites that were significantly altered in PMS participants with class II mutations and confirmed a general reduction in sphingolipid metabolism. Collectively, these results provide new evidence linking PMS participants carrying class II mutations with decreased expression of cytotoxic cell signatures, reduced relative proportions of NK cells, and lower sphingolipid metabolism. These findings highlight alternative avenues for therapeutic development and offer new mechanistic insights supporting genotype-to-phenotype associations in PMS.
Keywords: SHANK3; autism spectrum disorder; immunogenetics; immunophenotyping; multi-omics; rare disorders.
© 2022 The Authors.
Conflict of interest statement
A.K. receives research support from AMO Pharma and consults to Acadia, Alkermes, Neuren, and GW Pharma. He serves on scientific advisory boards for Ovid Therapeutics, Jaguar Therapeutics, and Ritrova Therapeutics. M.S. reports grant support from Novartis, Biogen, Astellas, Aeovian, Bridgebio, and Aucta. He has served on scientific advisory boards for Novartis, Roche, Regenxbio, and Alkermes. The remaining authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
