

A mechanistic framework for cardiometabolic and coronary artery diseases
- PMID: 36276926
- PMCID: PMC9583458
- DOI: 10.1038/s44161-021-00009-1
A mechanistic framework for cardiometabolic and coronary artery diseases
Abstract
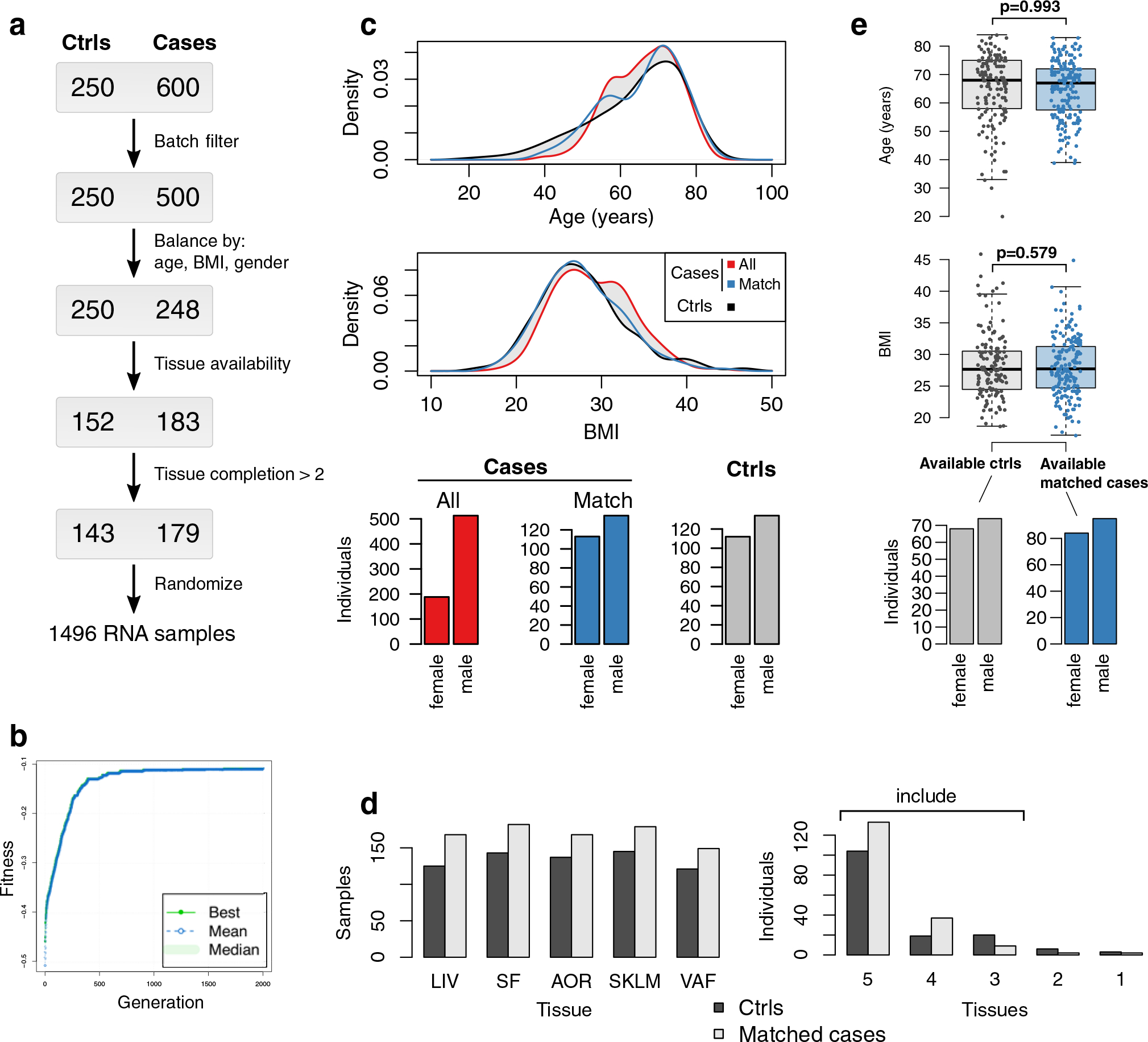
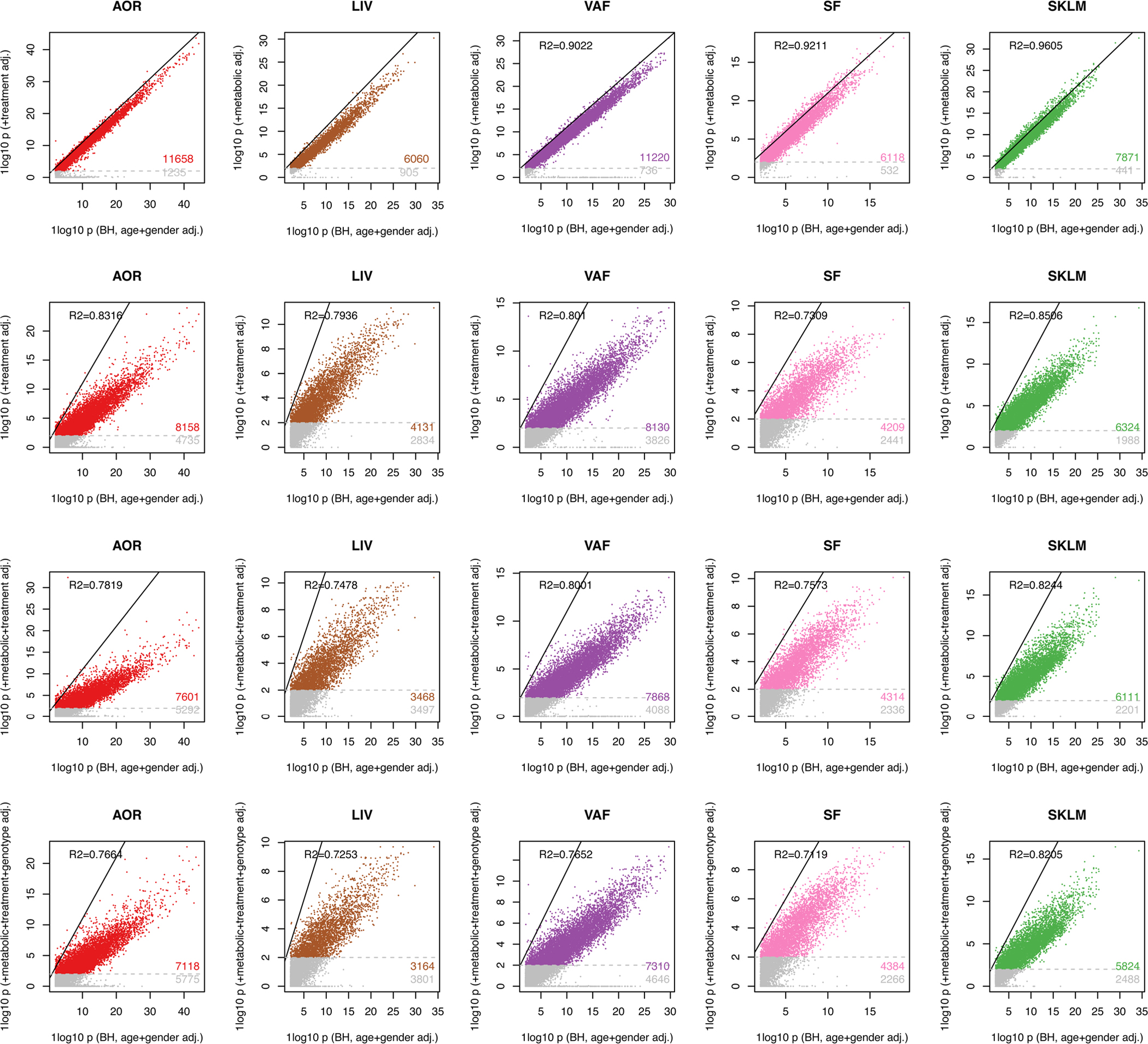
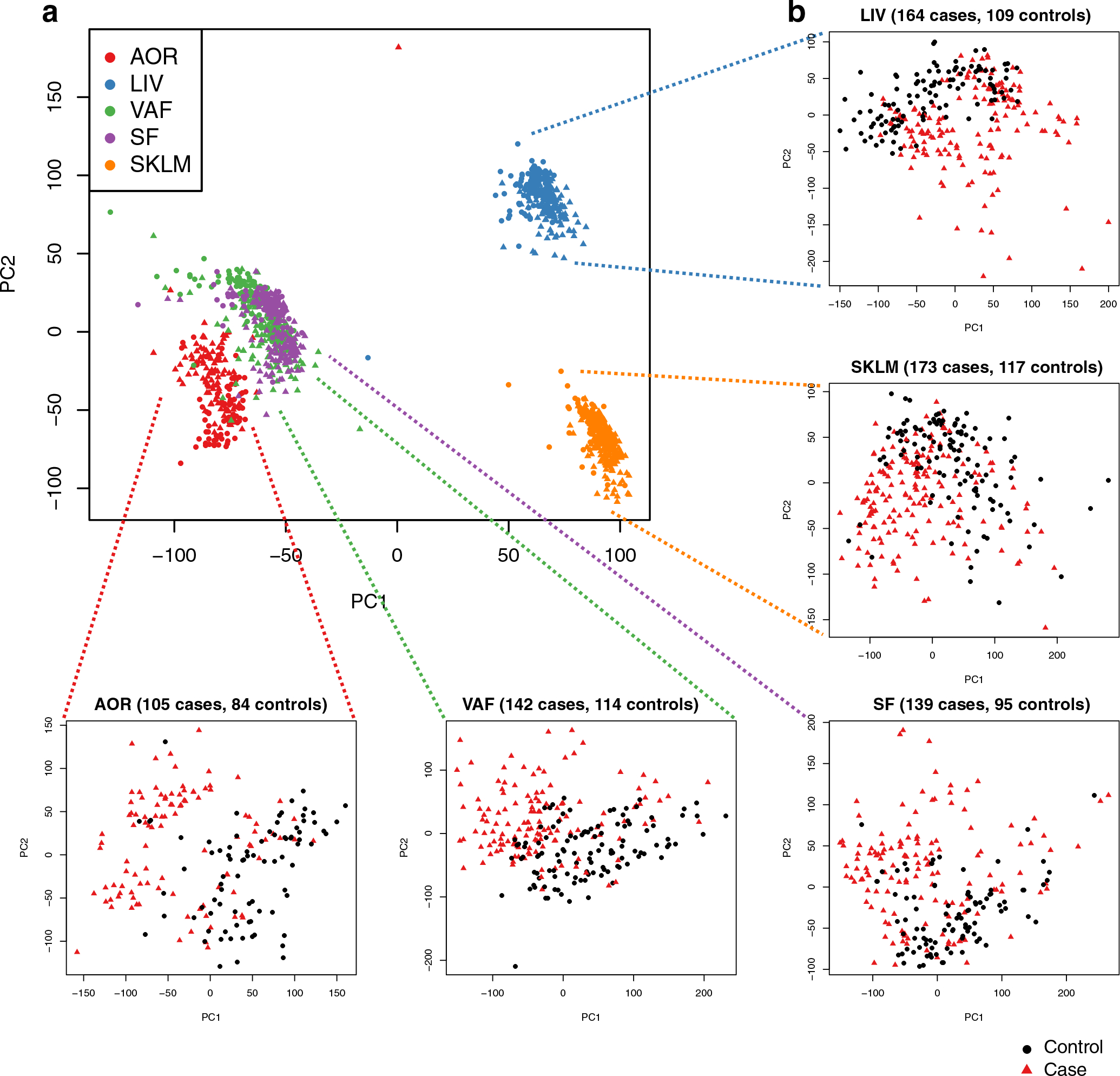
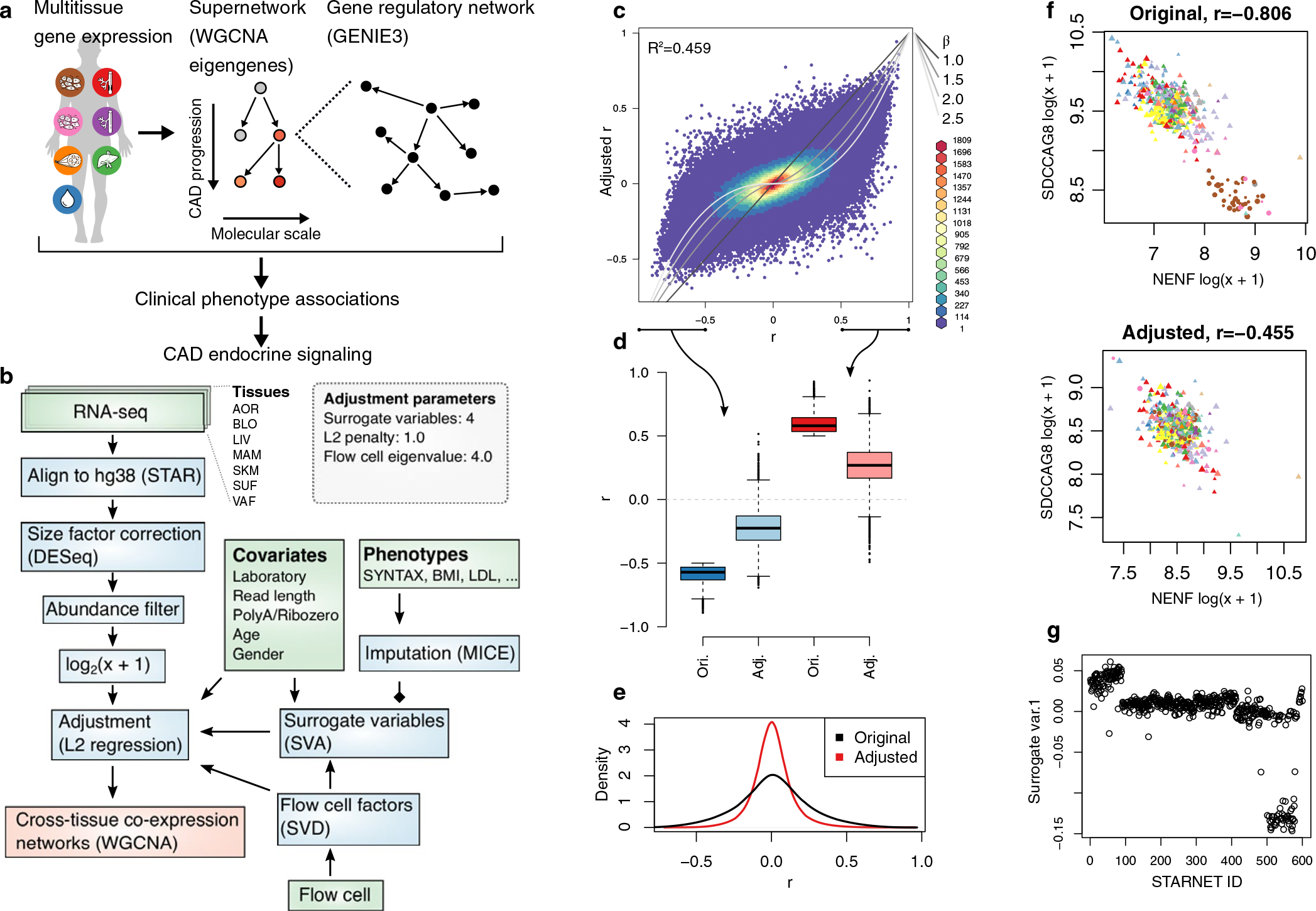
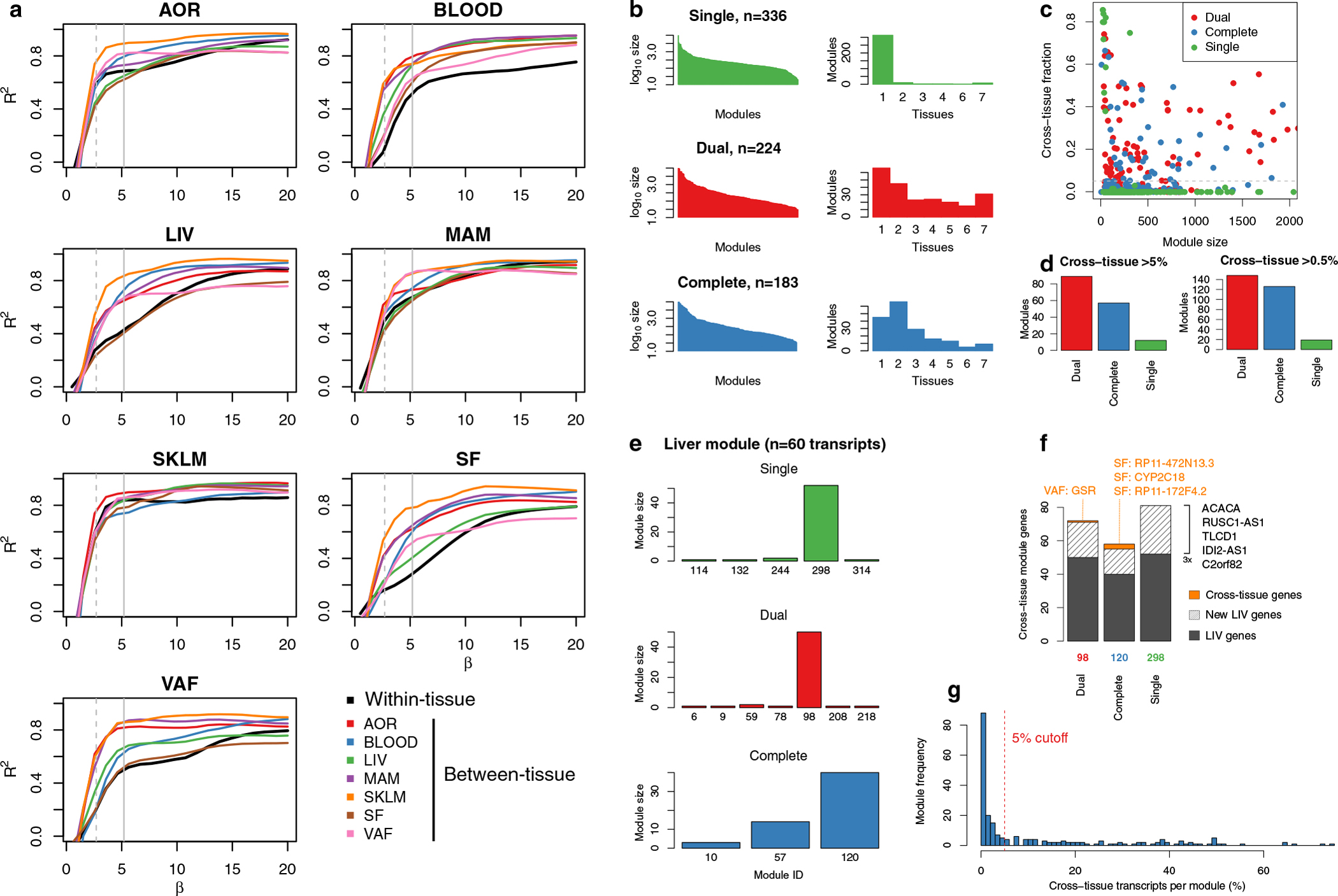
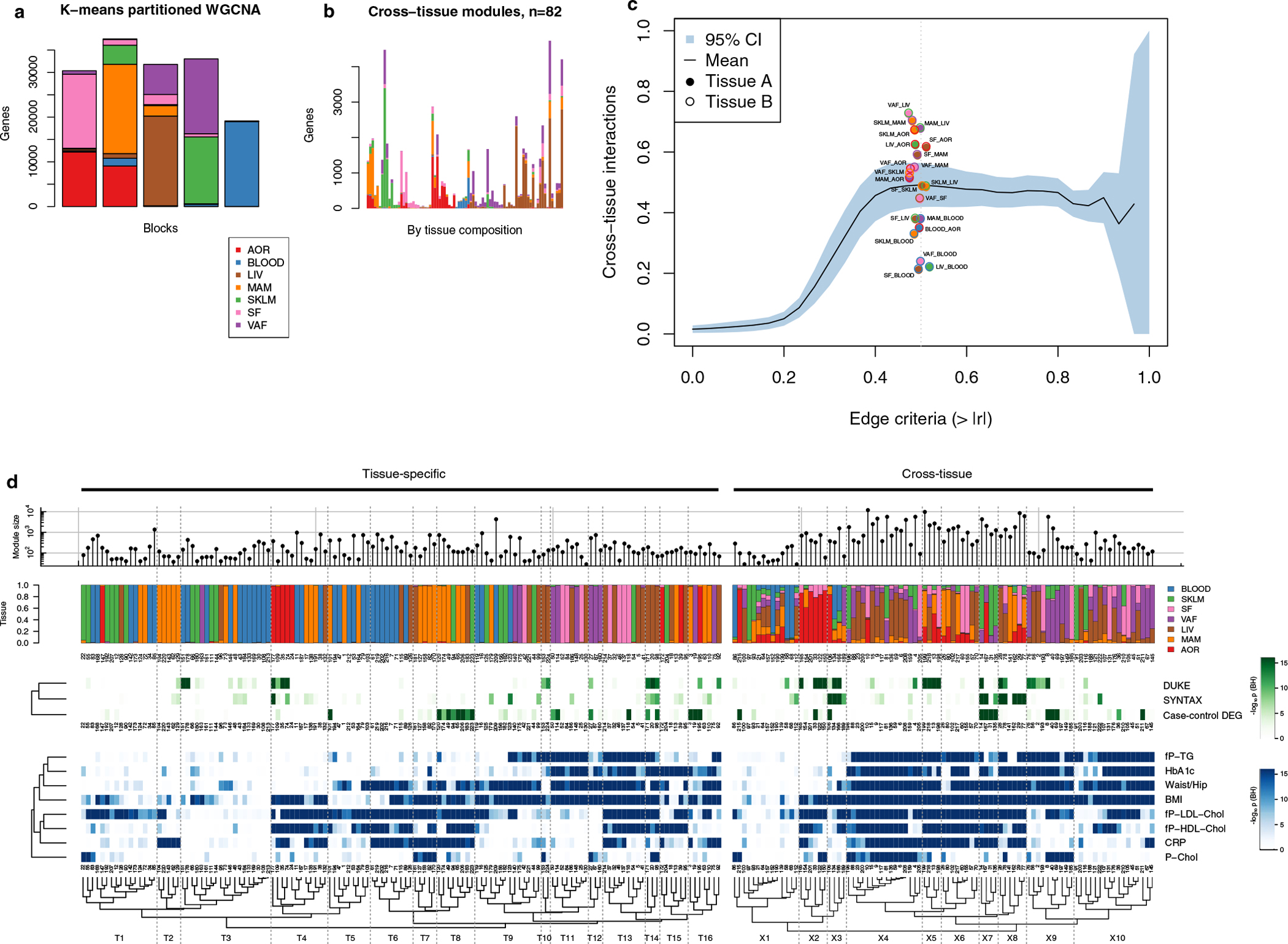
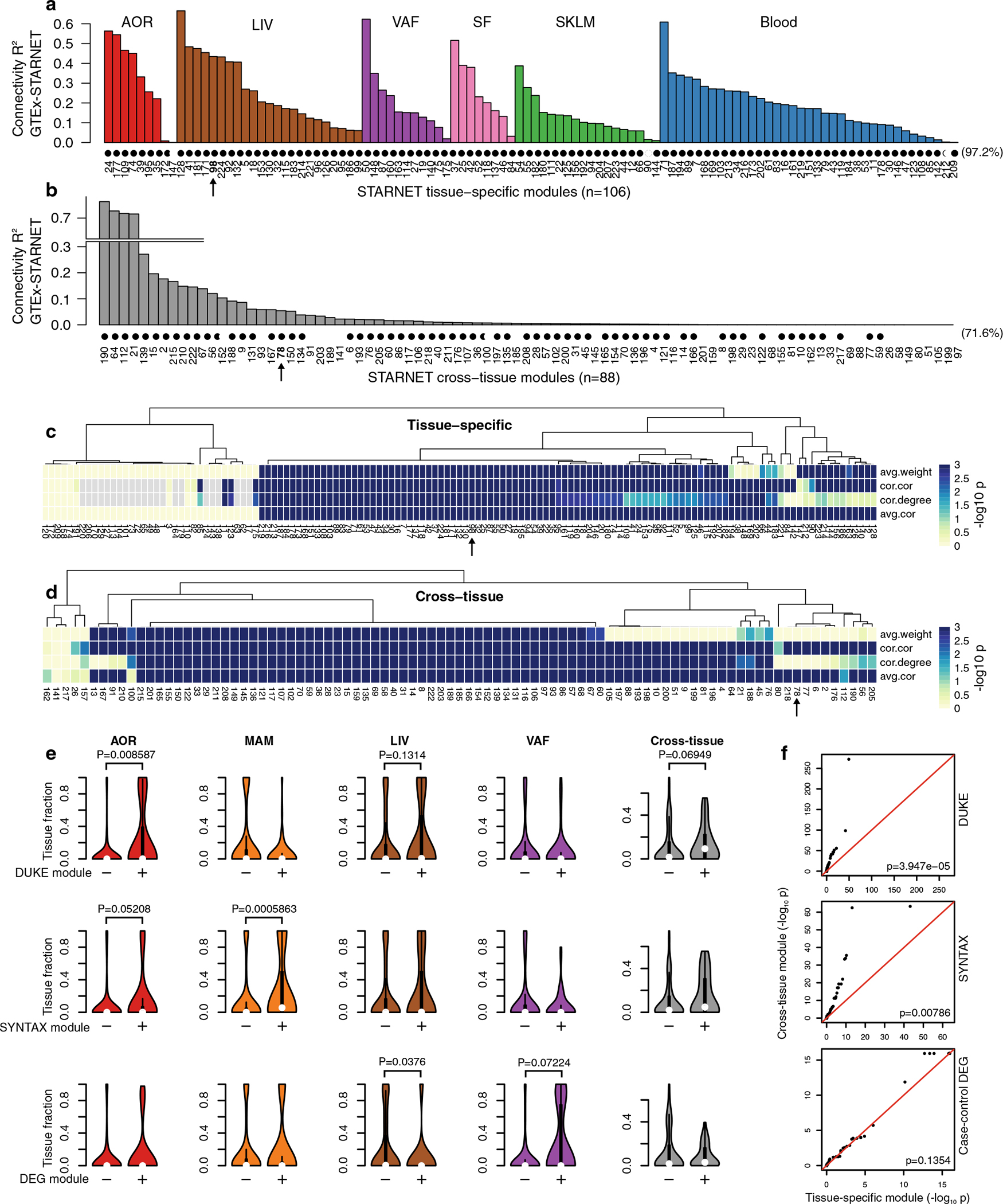
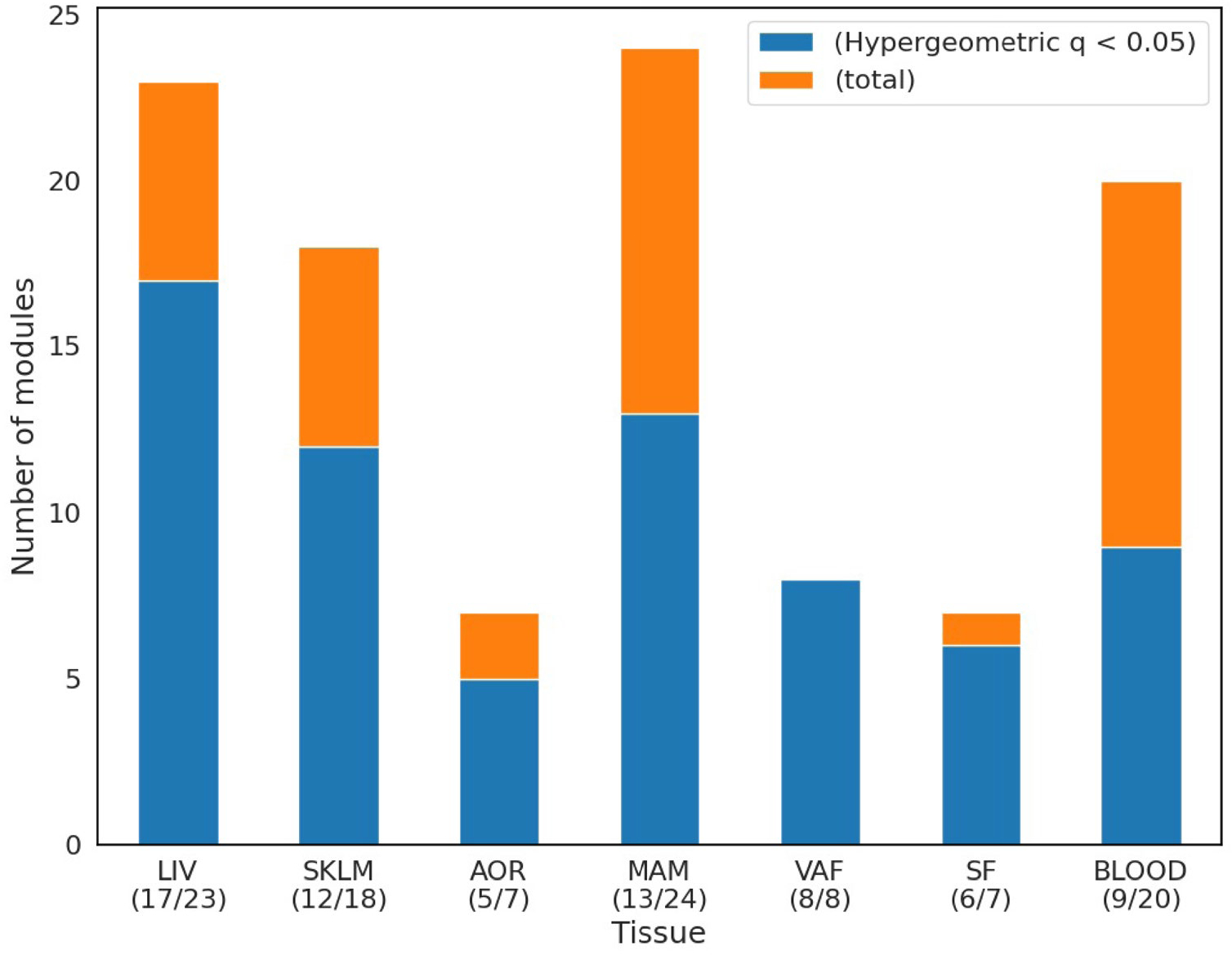
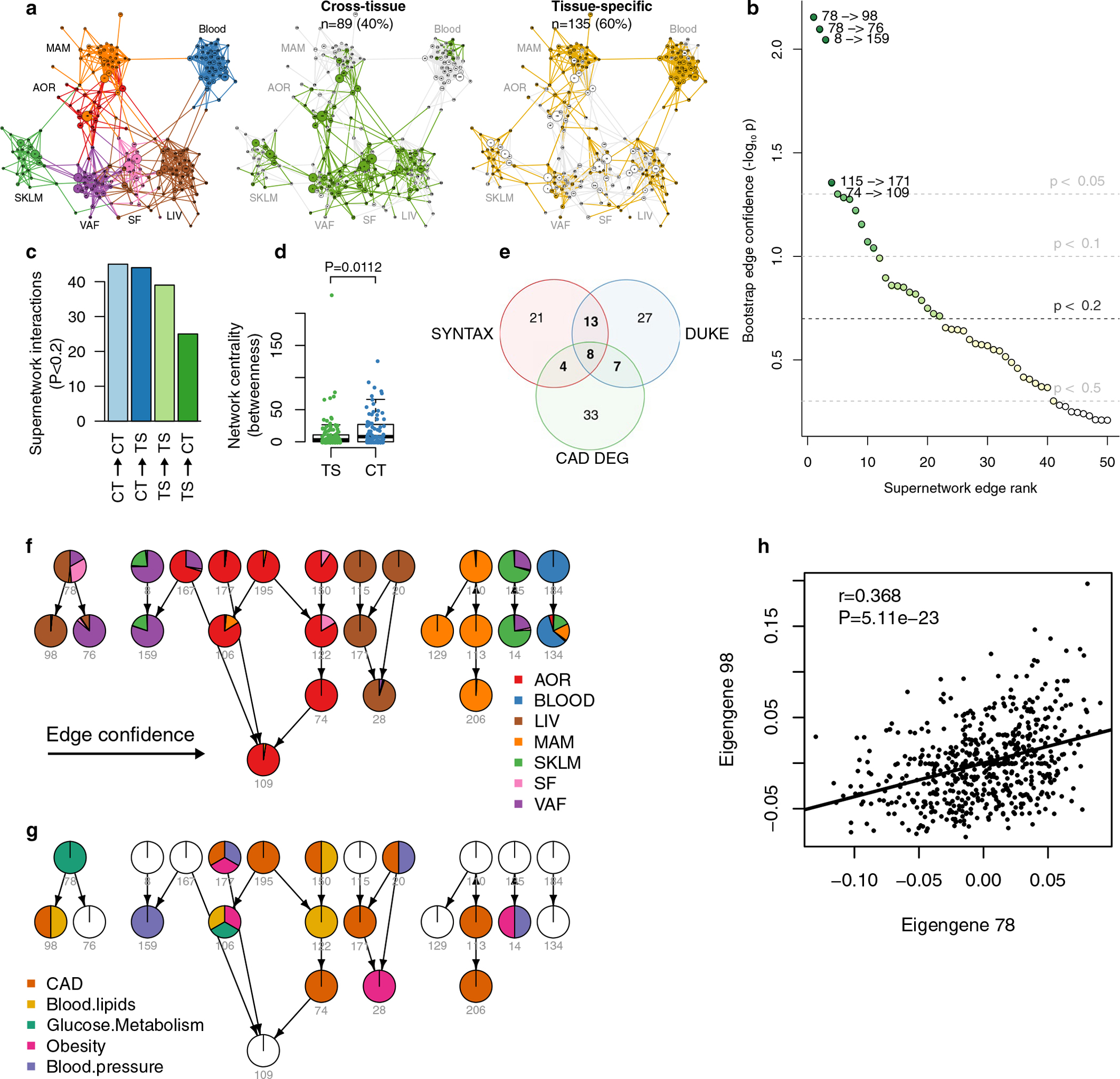
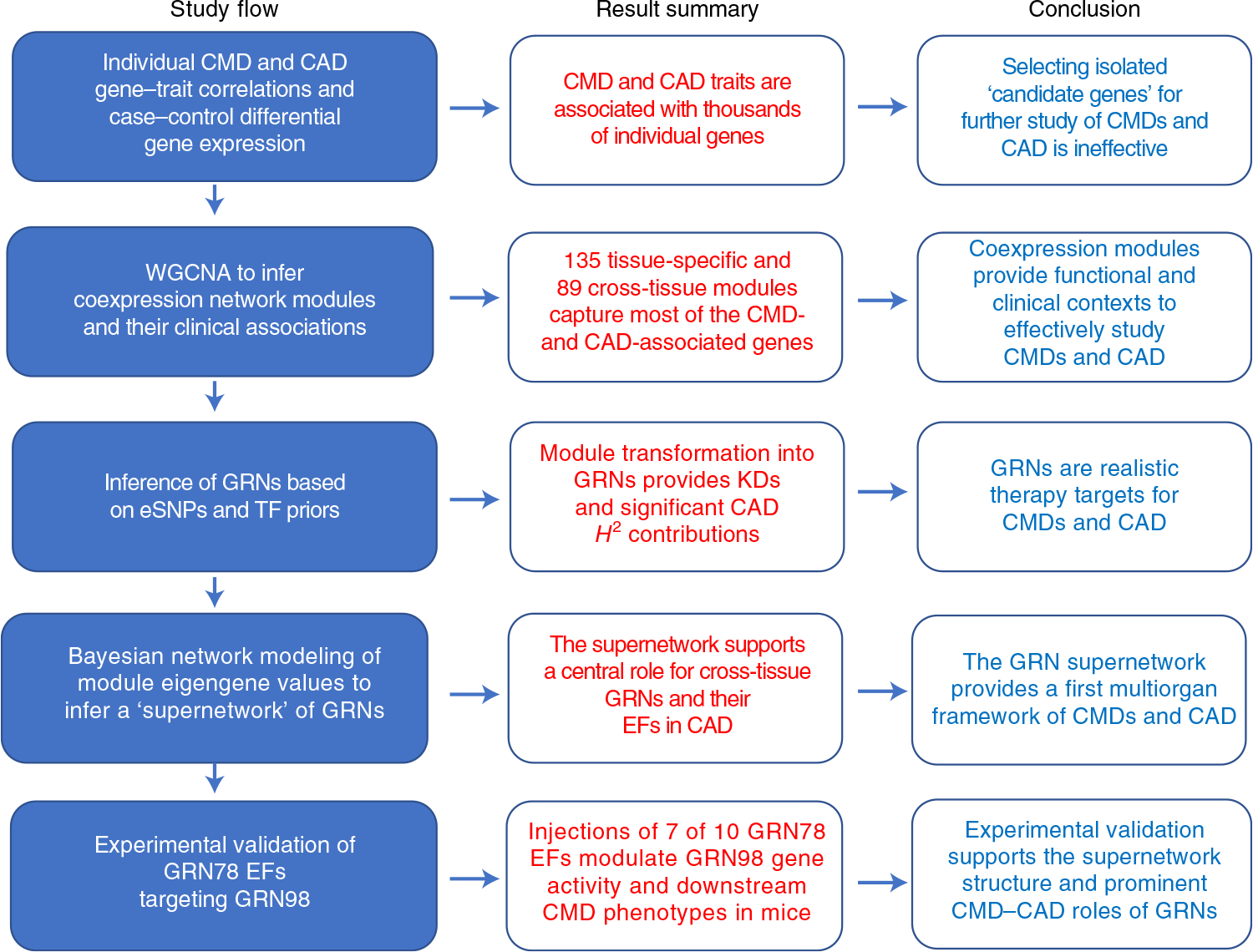
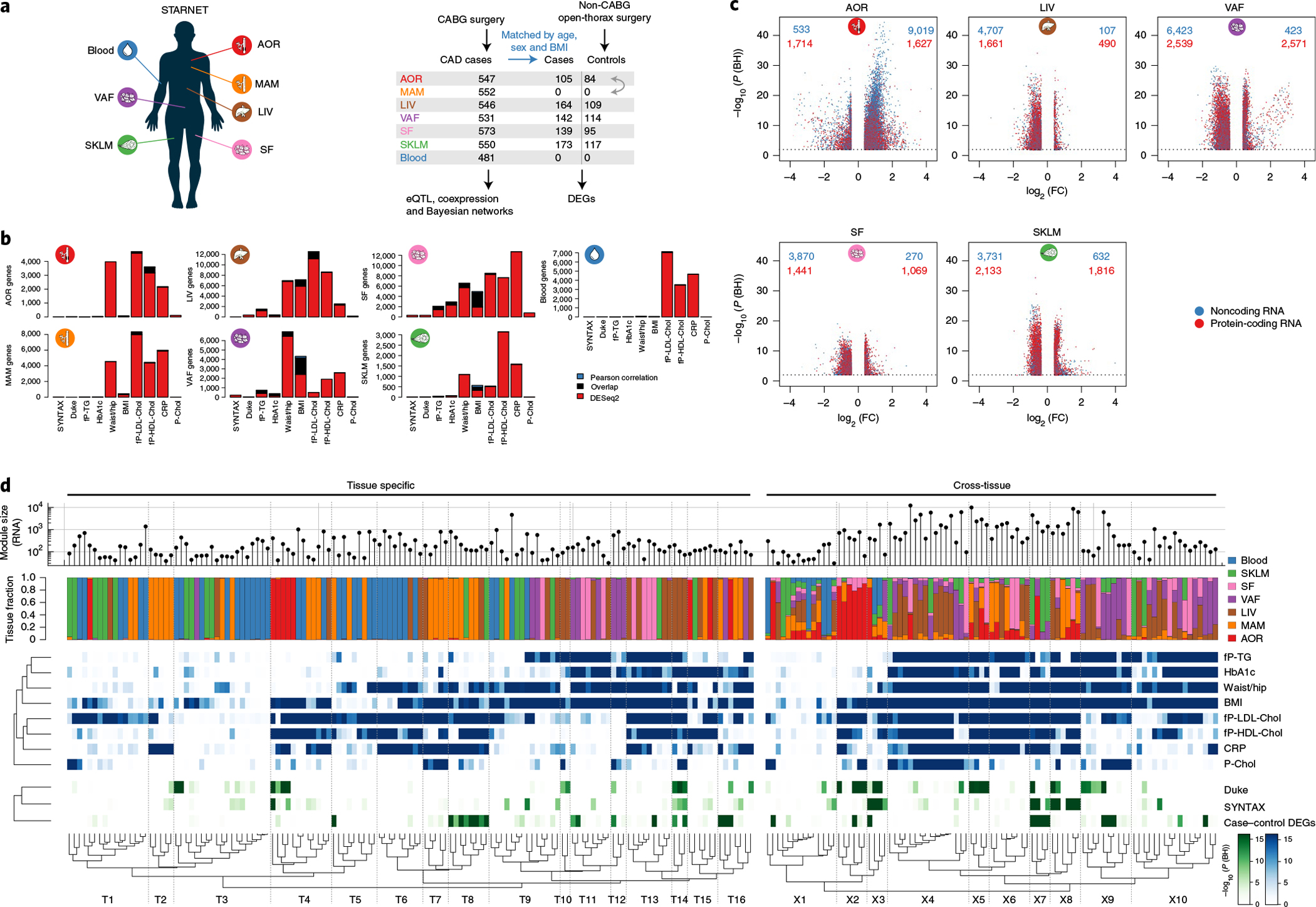
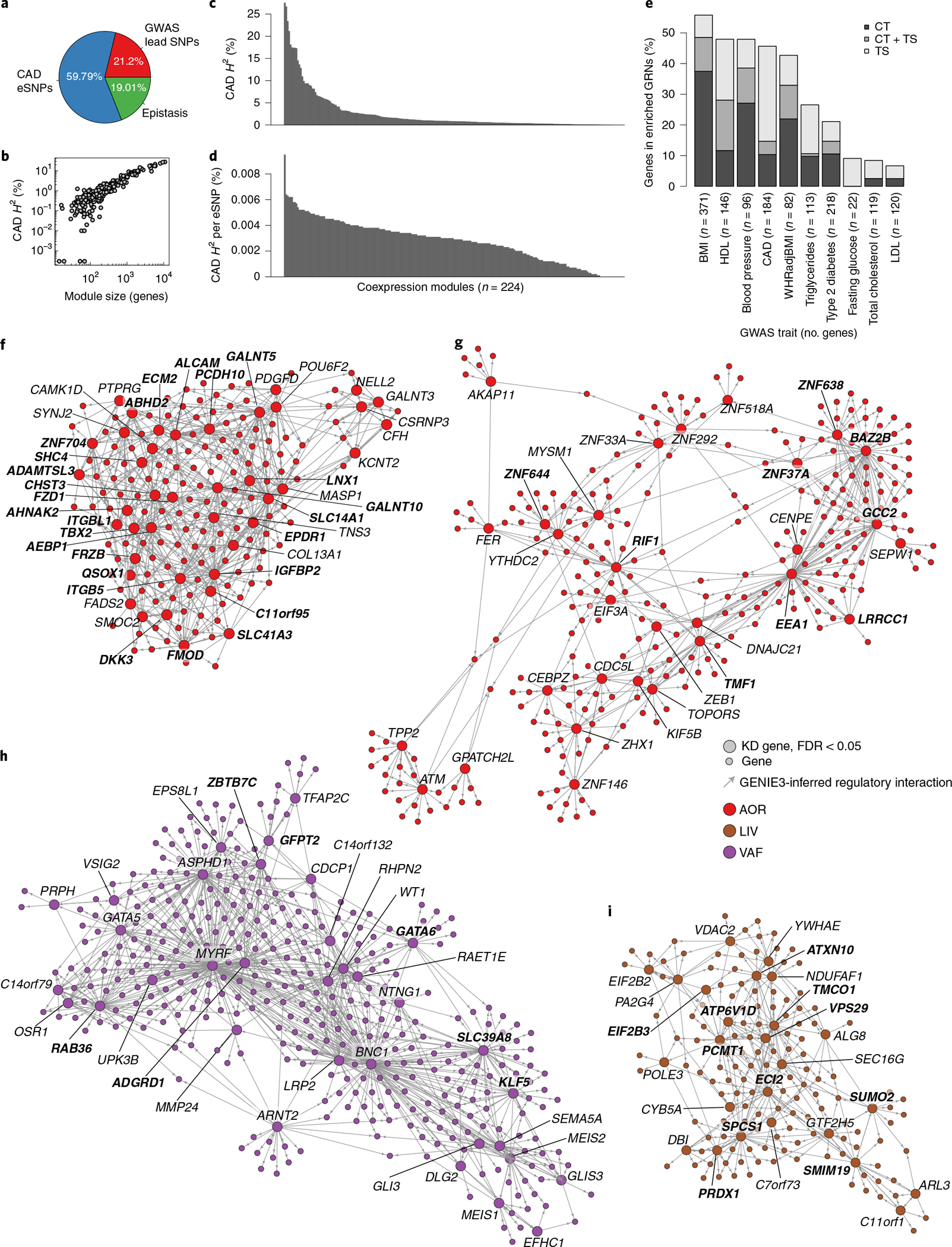
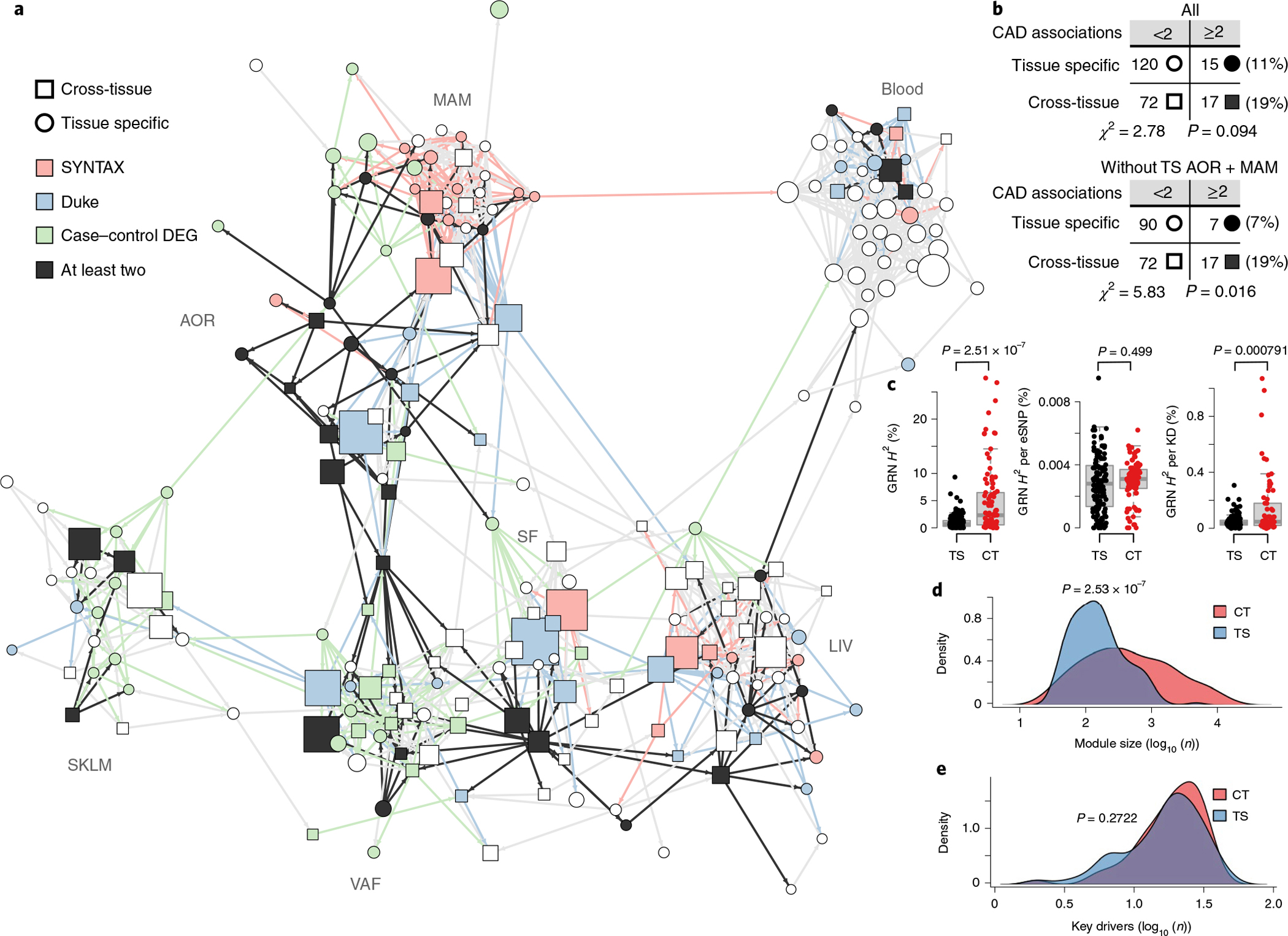
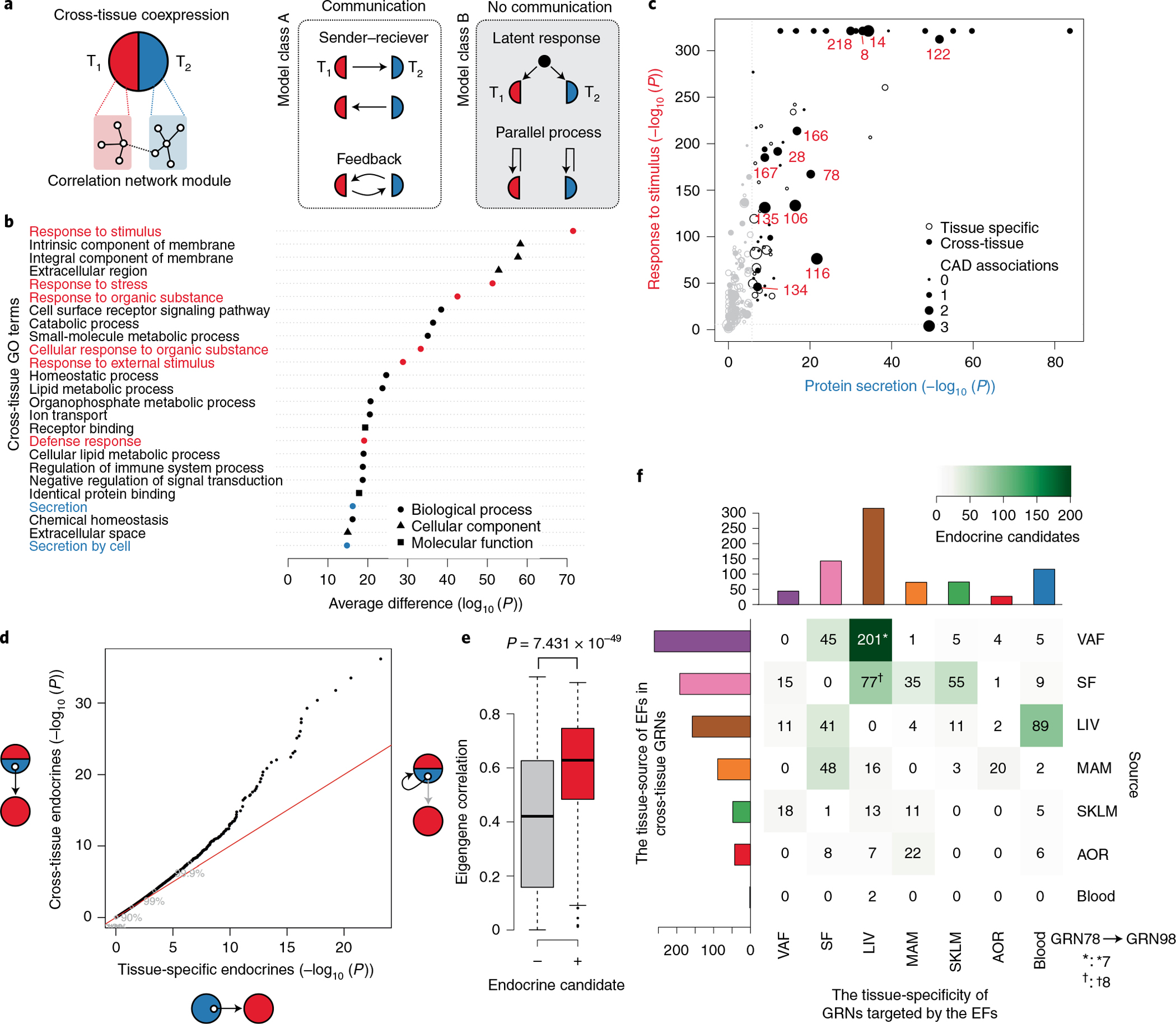
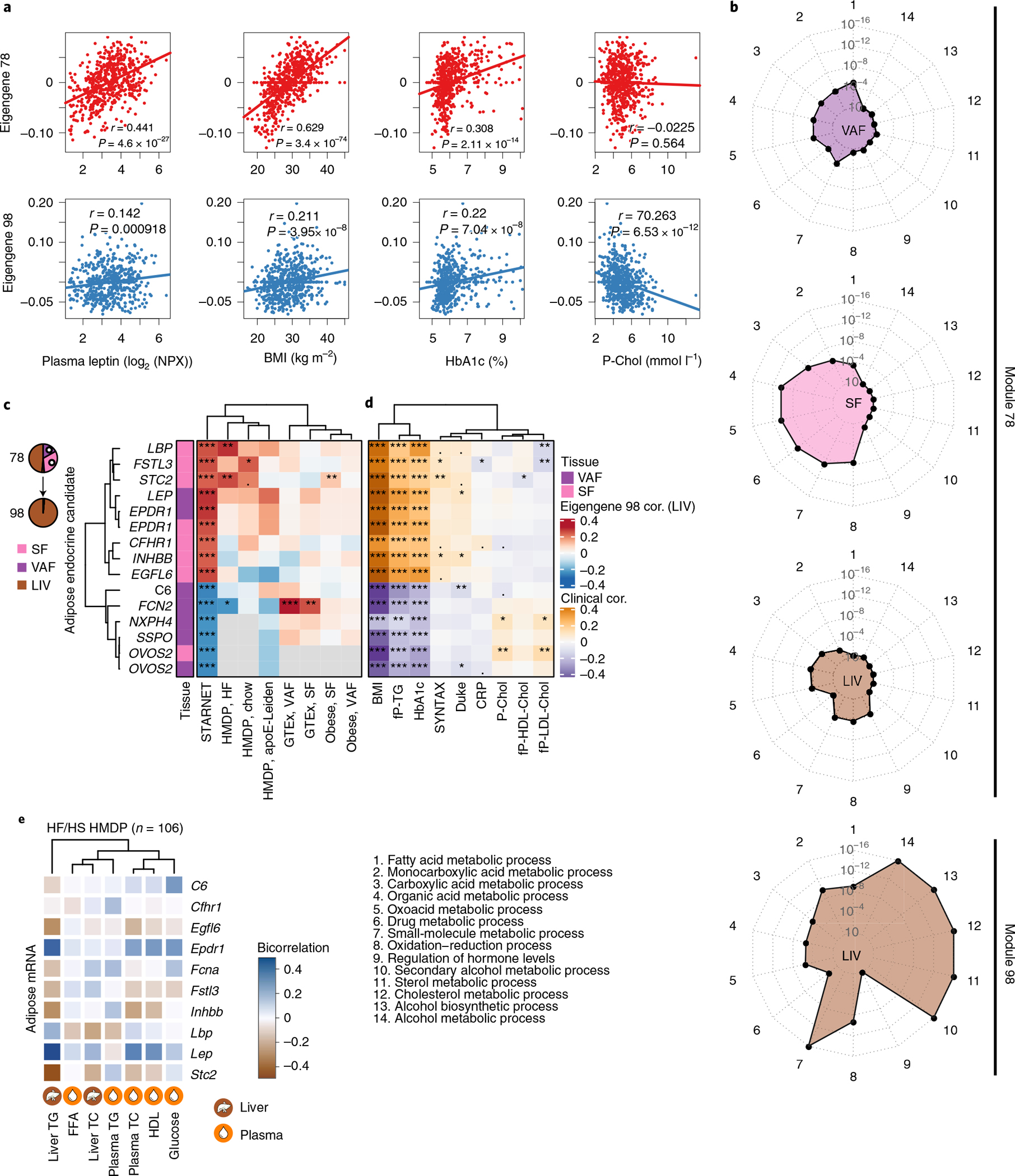
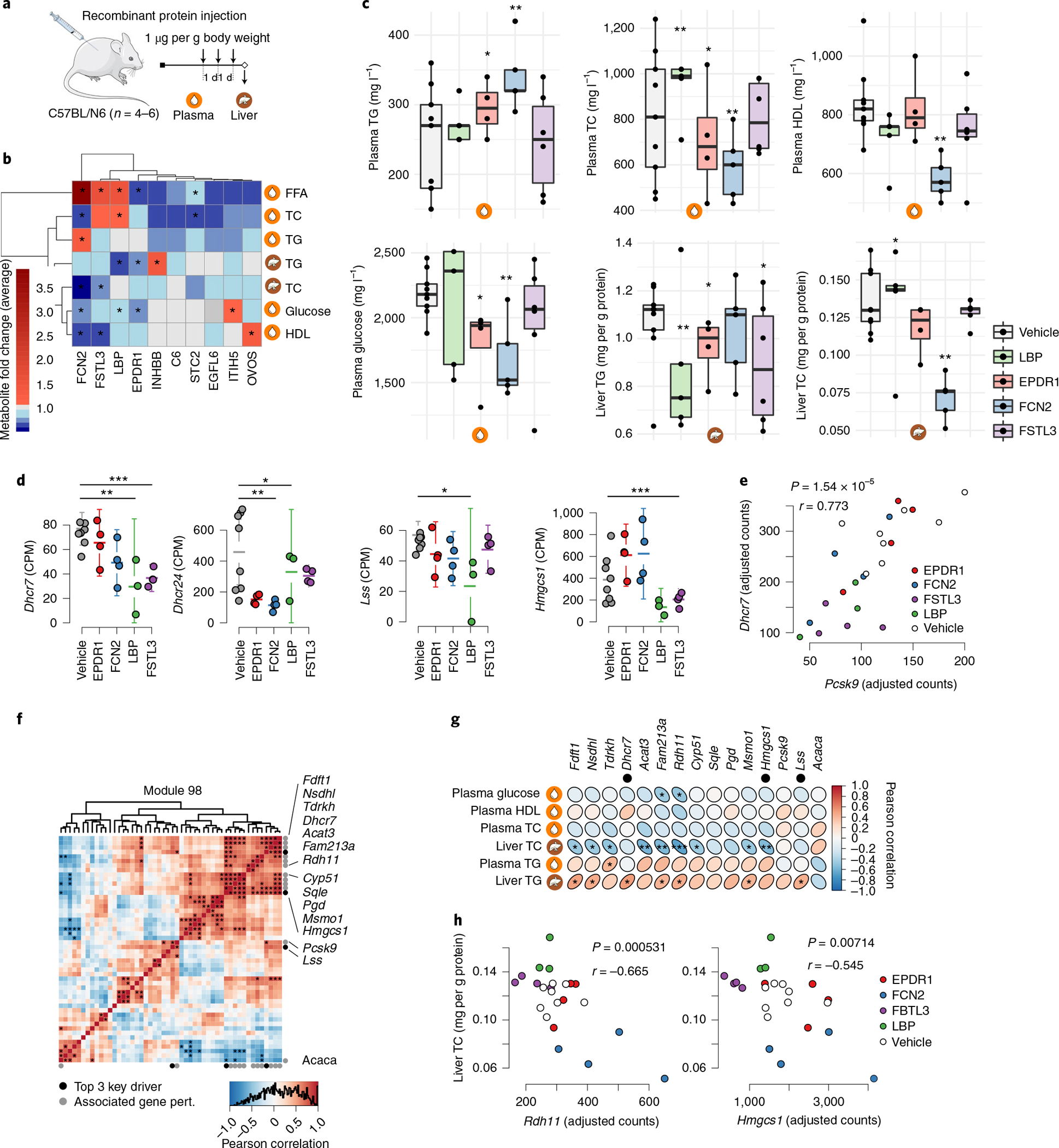
Coronary atherosclerosis results from the delicate interplay of genetic and exogenous risk factors, principally taking place in metabolic organs and the arterial wall. Here we show that 224 gene-regulatory coexpression networks (GRNs) identified by integrating genetic and clinical data from patients with (n = 600) and without (n = 250) coronary artery disease (CAD) with RNA-seq data from seven disease-relevant tissues in the Stockholm-Tartu Atherosclerosis Reverse Network Engineering Task (STARNET) study largely capture this delicate interplay, explaining >54% of CAD heritability. Within 89 cross-tissue GRNs associated with clinical severity of CAD, 374 endocrine factors facilitated inter-organ interactions, primarily along an axis from adipose tissue to the liver (n = 152). This axis was independently replicated in genetically diverse mouse strains and by injection of recombinant forms of adipose endocrine factors (EPDR1, FCN2, FSTL3 and LBP) that markedly altered blood lipid and glucose levels in mice. Altogether, the STARNET database and the associated GRN browser (http://starnet.mssm.edu) provide a multiorgan framework for exploration of the molecular interplay between cardiometabolic disorders and CAD.
Conflict of interest statement
Competing interests J.L.M.B. is the founder of Clinical Gene Networks (CGN). J.L.M.B. (chair) and A.R. are on CGN’s board of directors. J.L.M.B., A.R. and T.M. are shareholders in CGN. J.L.M.B. receives financial compensation as a consultant for CGN. CGN has an invested interest in STARNET that is regulated in an agreement with the Icahn School of Medicine at Mount Sinai. Neither the Icahn School of Medicine at Mount Sinai nor CGN have made claims to results presented in this study. E.E.S. is the CEO of Sema4. C.W. and L.-M.G. are employees of AstraZeneca. No funding for this study was received from Sema4. AstraZeneca supported this study through independent grants to J.L.M.B. at the Karolinska Institutet (ICMC). The remaining authors declare no competing interests.
Figures

References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous