

A CRISPR/Cas12a-responsive dual-aptamer DNA network for specific capture and controllable release of circulating tumor cells
- PMID: 36277631
- PMCID: PMC9473531
- DOI: 10.1039/d2sc03374g
A CRISPR/Cas12a-responsive dual-aptamer DNA network for specific capture and controllable release of circulating tumor cells
Abstract
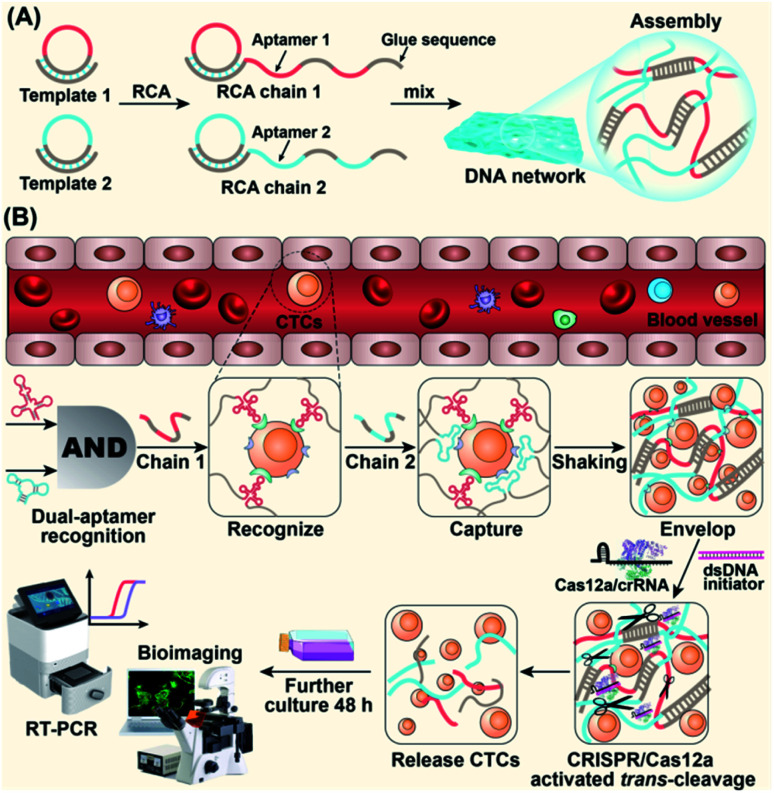
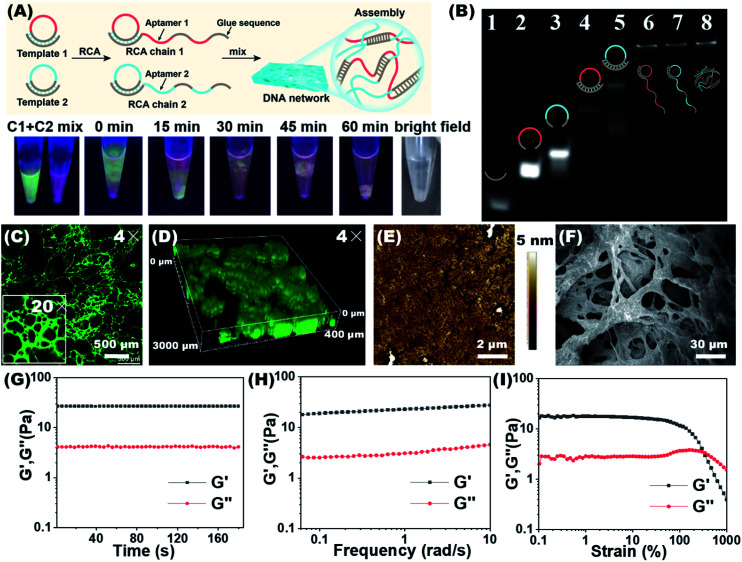
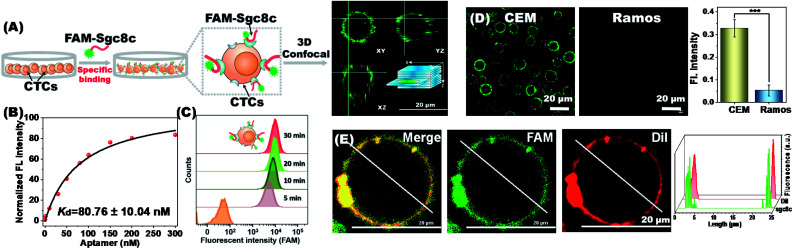
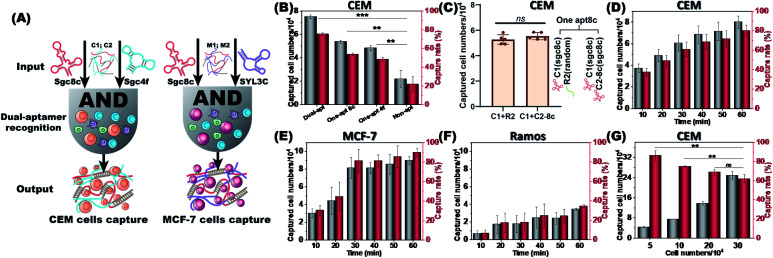
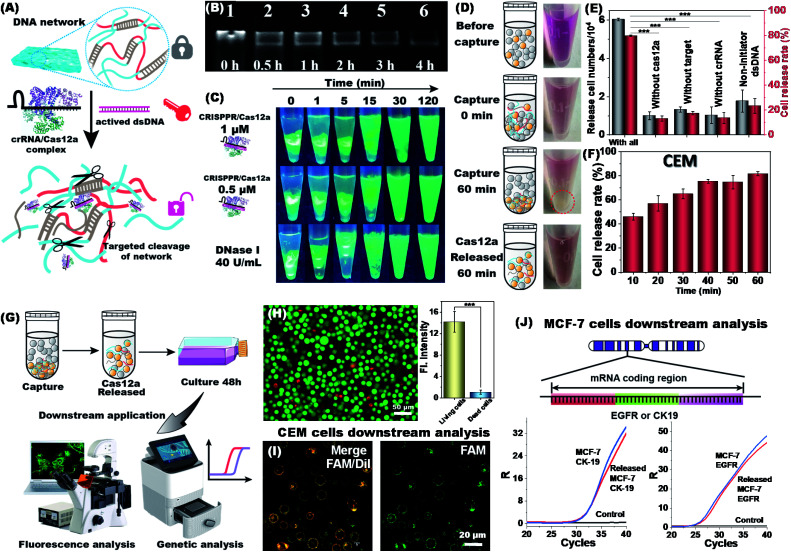
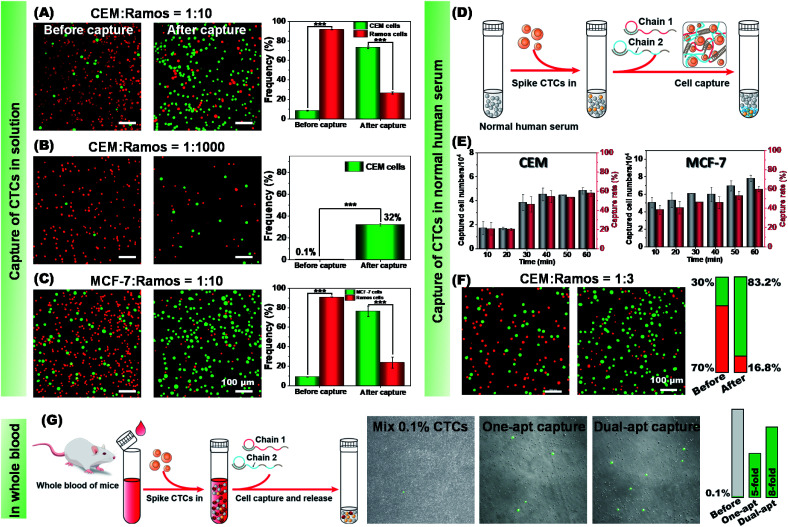
The separation and detection of circulating tumor cells (CTCs) have a significant impact on clinical diagnosis and treatment by providing a predictive diagnosis of primary tumors and tumor metastasis. But the responsive release and downstream analysis of live CTCs will provide more valuable information about molecular markers and functional properties. To this end, specific capture and controllable release methods, which can achieve the highly efficient enrichment of CTCs with strong viability, are urgently needed. DNA networks create a flexible, semi-wet three-dimensional (3D) microenvironment for cell culture, and have the potential to minimize the loss of cell viability and molecular integrity. More importantly, responsive DNA networks can be reasonably designed as smart sensors and devices to change shape, color, disassemble, and giving back to external stimuli. Here, a strategy for specifically collecting cells using a dual-aptamer DNA network is designed. The proposed strategy enables effective capture, 3D encapsulation, and responsive release of CTCs with strong viability, which can be used for downstream analysis of live cells. The programmability of CRISPR/Cas12a, a powerful toolbox for genome editing, is used to activate the responsive release of captured CTCs from the DNA network. After activation by a specified double-strand DNA (dsDNA) input, CRISPR/Cas12a cleaves the single-stranded DNA regions in the network, resulting in molecular to macroscopic changes in the network. Accompanied by the deconstruction of the DNA network into fragments, controllable cell release is achieved. The viability of released CTCs is well maintained and downstream cell analysis can be performed. This strategy uses the enzymatic properties of CRISPR/Cas12a to design a platform to improve the programmability and versatility of the DNA network, providing a powerful and effective method for capturing and releasing CTCs from complex physiological samples.
This journal is © The Royal Society of Chemistry.
Conflict of interest statement
There are no conflicts to declare.
Figures

Similar articles
-
Multivalent Duplexed-Aptamer Networks Regulated a CRISPR-Cas12a System for Circulating Tumor Cell Detection.Anal Chem. 2021 Sep 28;93(38):12921-12929. doi: 10.1021/acs.analchem.1c02228. Epub 2021 Sep 17. Anal Chem. 2021. PMID: 34533940
-
Tumor Microenvironment-Inspired Glutathione-Responsive Three-Dimensional Fibrous Network for Efficient Trapping and Gentle Release of Circulating Tumor Cells.ACS Appl Mater Interfaces. 2023 May 24;15(20):24013-24022. doi: 10.1021/acsami.3c00307. Epub 2023 May 13. ACS Appl Mater Interfaces. 2023. PMID: 37178127
-
Aptamer-mediated DNA concatemer functionalized magnetic nanoparticles for reversible capture and release of circulating tumor cells.Colloids Surf B Biointerfaces. 2022 Oct;218:112733. doi: 10.1016/j.colsurfb.2022.112733. Epub 2022 Jul 28. Colloids Surf B Biointerfaces. 2022. PMID: 35981473
-
Development of Metal-Organic Framework-Based Dual Antibody Nanoparticles for the Highly Specific Capture and Gradual Release of Circulating Tumor Cells.Front Bioeng Biotechnol. 2022 Feb 7;10:806238. doi: 10.3389/fbioe.2022.806238. eCollection 2022. Front Bioeng Biotechnol. 2022. PMID: 35198544 Free PMC article. Review.
-
Nanostructure embedded microchips for detection, isolation, and characterization of circulating tumor cells.Acc Chem Res. 2014 Oct 21;47(10):2941-50. doi: 10.1021/ar5001617. Epub 2014 Aug 11. Acc Chem Res. 2014. PMID: 25111636 Free PMC article. Review.
Cited by
-
Photo-functionalized TiO2 film for facile immobilization of EpCAM antibodies and efficient enrichment of circulating tumor cells.Front Pharmacol. 2023 Feb 28;14:1126602. doi: 10.3389/fphar.2023.1126602. eCollection 2023. Front Pharmacol. 2023. PMID: 36925636 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Other Literature Sources