

Congenital aniridia beyond black eyes: From phenotype and novel genetic mechanisms to innovative therapeutic approaches
- PMID: 36280537
- PMCID: PMC11062406
- DOI: 10.1016/j.preteyeres.2022.101133
Congenital aniridia beyond black eyes: From phenotype and novel genetic mechanisms to innovative therapeutic approaches
Abstract
Congenital PAX6-aniridia, initially characterized by the absence of the iris, has progressively been shown to be associated with other developmental ocular abnormalities and systemic features making congenital aniridia a complex syndromic disorder rather than a simple isolated disease of the iris. Moreover, foveal hypoplasia is now recognized as a more frequent feature than complete iris hypoplasia and a major visual prognosis determinant, reversing the classical clinical picture of this disease. Conversely, iris malformation is also a feature of various anterior segment dysgenesis disorders caused by PAX6-related developmental genes, adding a level of genetic complexity for accurate molecular diagnosis of aniridia. Therefore, the clinical recognition and differential genetic diagnosis of PAX6-related aniridia has been revealed to be much more challenging than initially thought, and still remains under-investigated. Here, we update specific clinical features of aniridia, with emphasis on their genotype correlations, as well as provide new knowledge regarding the PAX6 gene and its mutational spectrum, and highlight the beneficial utility of clinically implementing targeted Next-Generation Sequencing combined with Whole-Genome Sequencing to increase the genetic diagnostic yield of aniridia. We also present new molecular mechanisms underlying aniridia and aniridia-like phenotypes. Finally, we discuss the appropriate medical and surgical management of aniridic eyes, as well as innovative therapeutic options. Altogether, these combined clinical-genetic approaches will help to accelerate time to diagnosis, provide better determination of the disease prognosis and management, and confirm eligibility for future clinical trials or genetic-specific therapies.
Keywords: Congenital aniridia; Foveal hypoplasia; Gene therapy; Next-generation sequencing; PAX6; Whole-genome sequencing.
Copyright © 2022. Published by Elsevier Ltd.
Conflict of interest statement
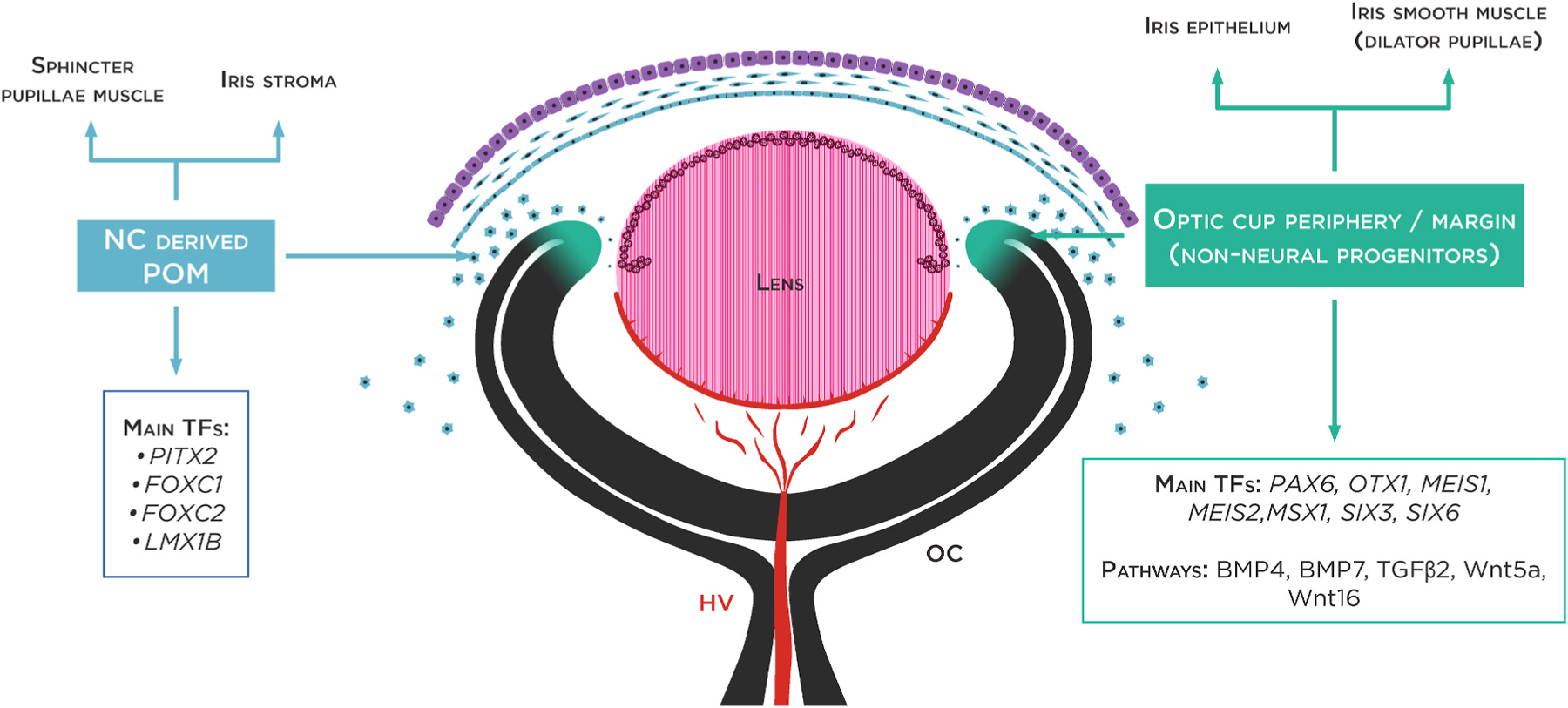
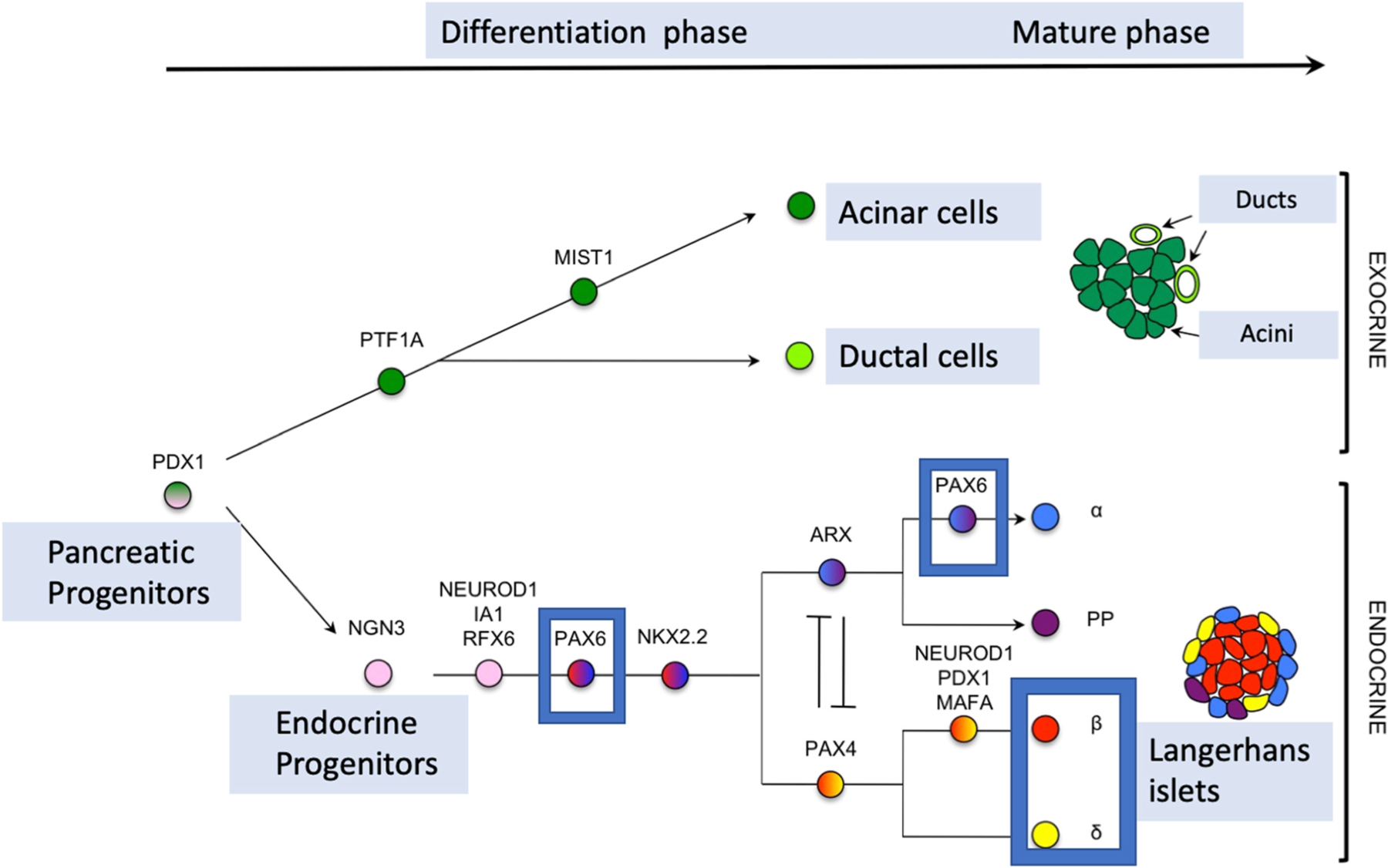
Declarations of competing interest None. The three layers of the cornea are defined: the corneal endothelium (CEnd) deriving from the first set of neural crest cells/POM cells, the corneal stroma (CS) deriving from the second wave of NC cells/POM cells and the corneal epithelium (CE) developing from the outer surface ectoderm. The third wave of NC cells/POM cells arrives at the angle of the future cornea and the periphery of the optic cup and contributes to pupillary membrane and iris stroma development. From the periphery of the optic cup will then originate the pigmented epithelium and the smooth muscle of the iris. HV (hyaloid vessels, tunica vasculosa lentis).
Figures
References
-
- Aalfs CM, Fantes JA, Wenniger-Prick LJ, Sluijter S, Hennekam RC, van Heyningen V, Hoovers JM, 1997. Tandem duplication of 11p12-p13 in a child with borderline development delay and eye abnormalities: dose effect of the PAX6 gene product? Am. J. Med. Genet. 73, 267–271. 10.1002/(sici)1096-8628(19971219)73:3<267::aid-ajmg7>3.0.co, 2. - DOI - PubMed
