

Spatial Transcriptomic Analysis of a Diverse Patient Cohort Reveals a Conserved Architecture in Triple-Negative Breast Cancer
- PMID: 36283023
- PMCID: PMC9812886
- DOI: 10.1158/0008-5472.CAN-22-2682
Spatial Transcriptomic Analysis of a Diverse Patient Cohort Reveals a Conserved Architecture in Triple-Negative Breast Cancer
Abstract
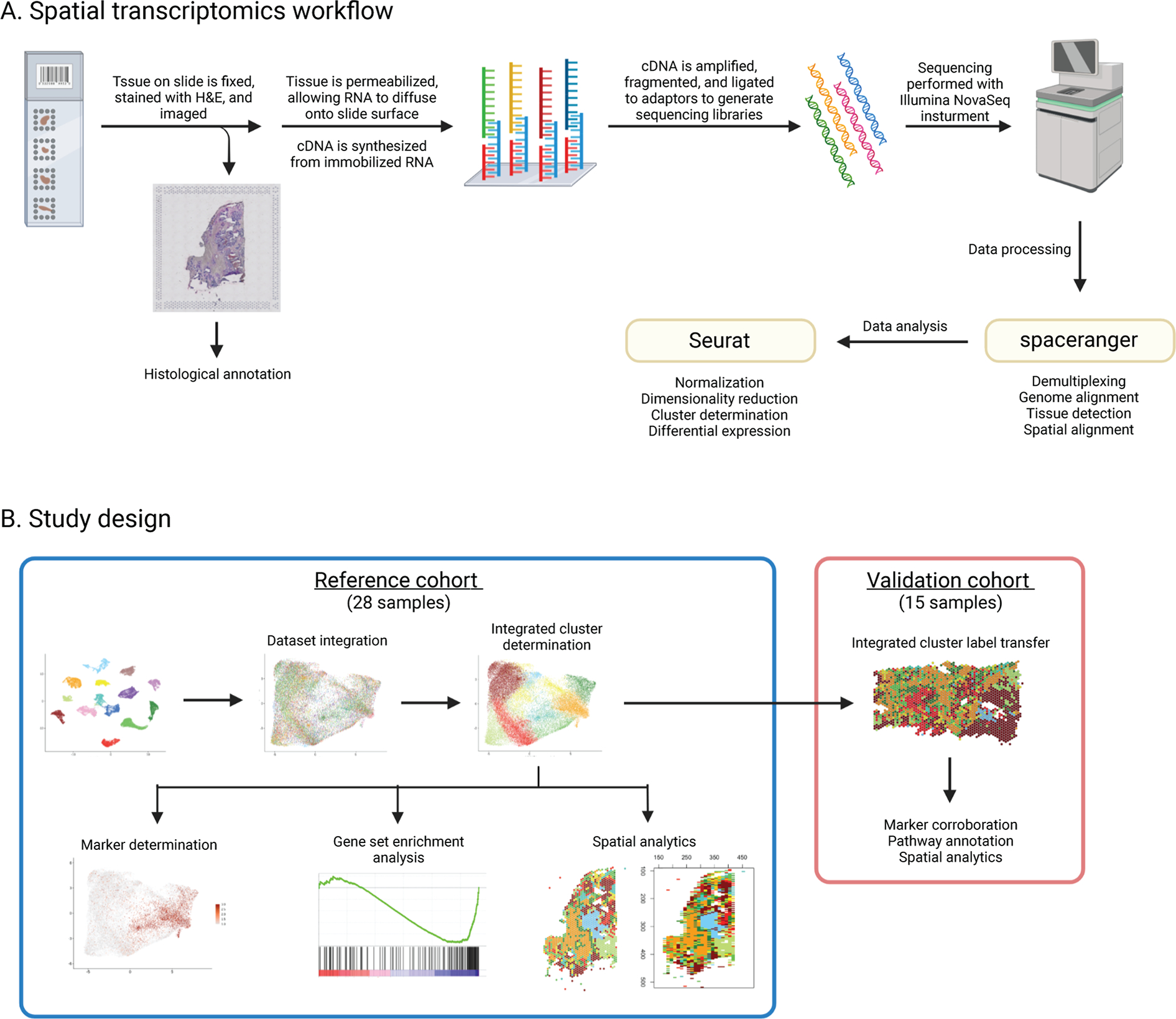
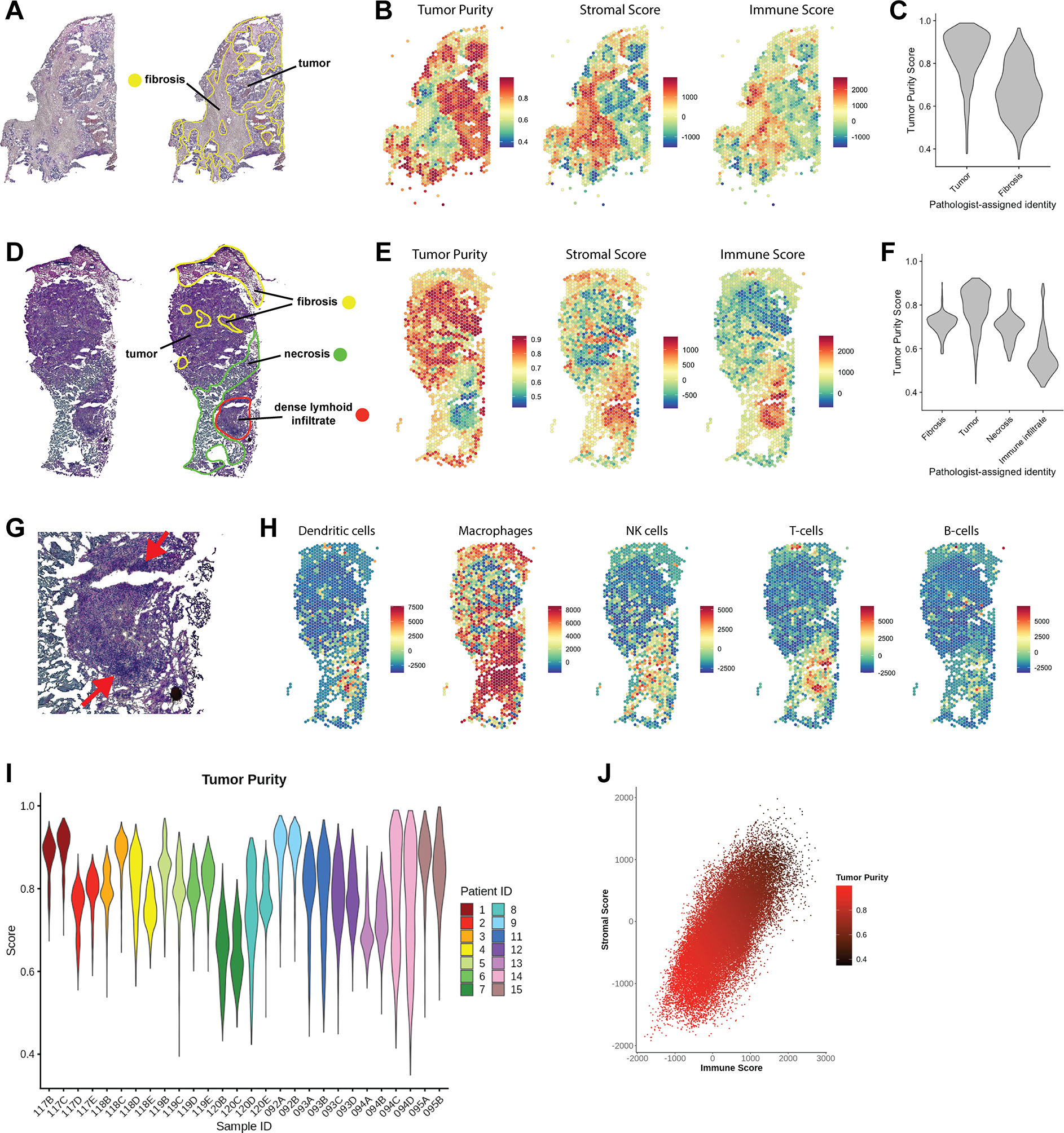
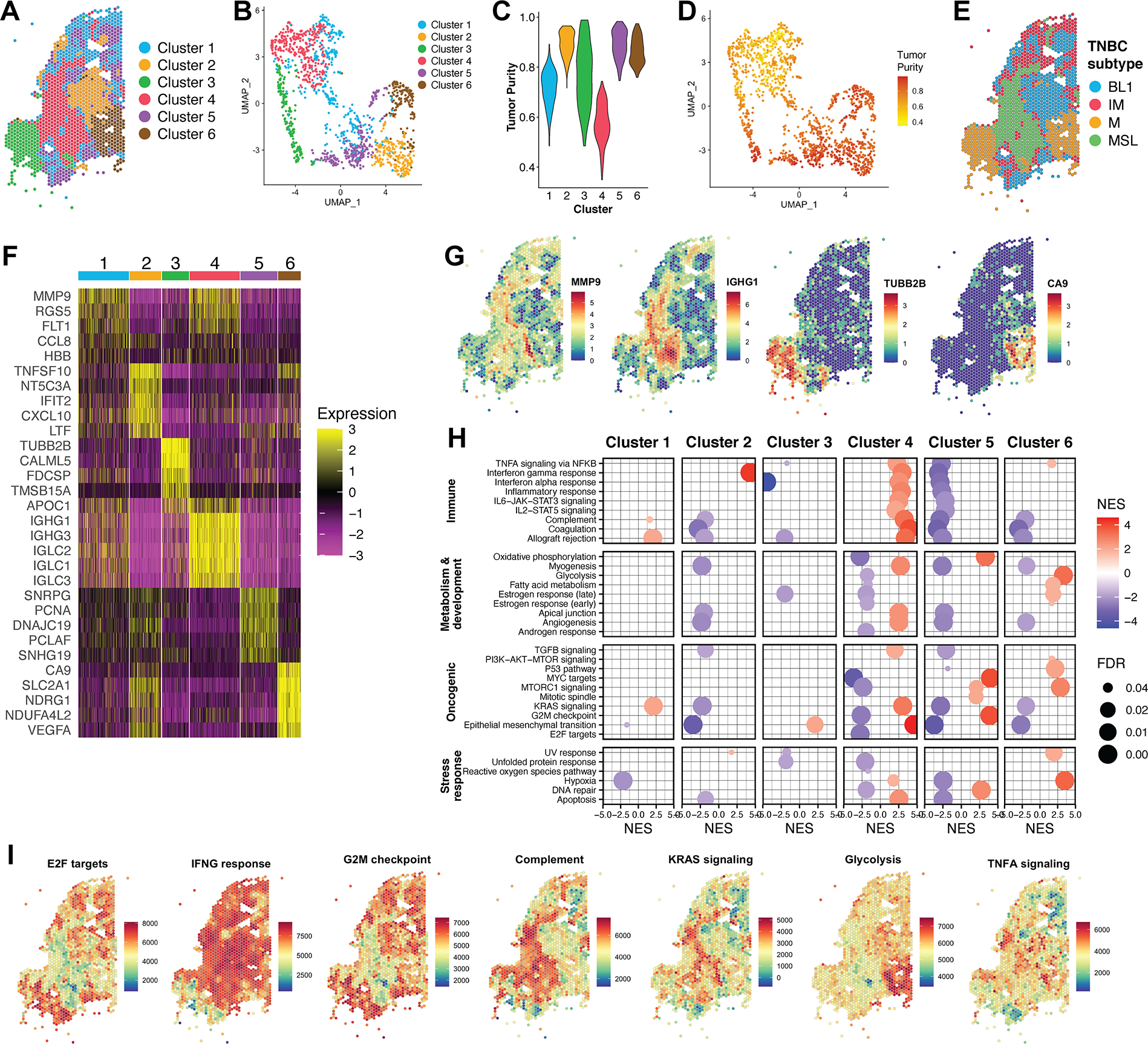
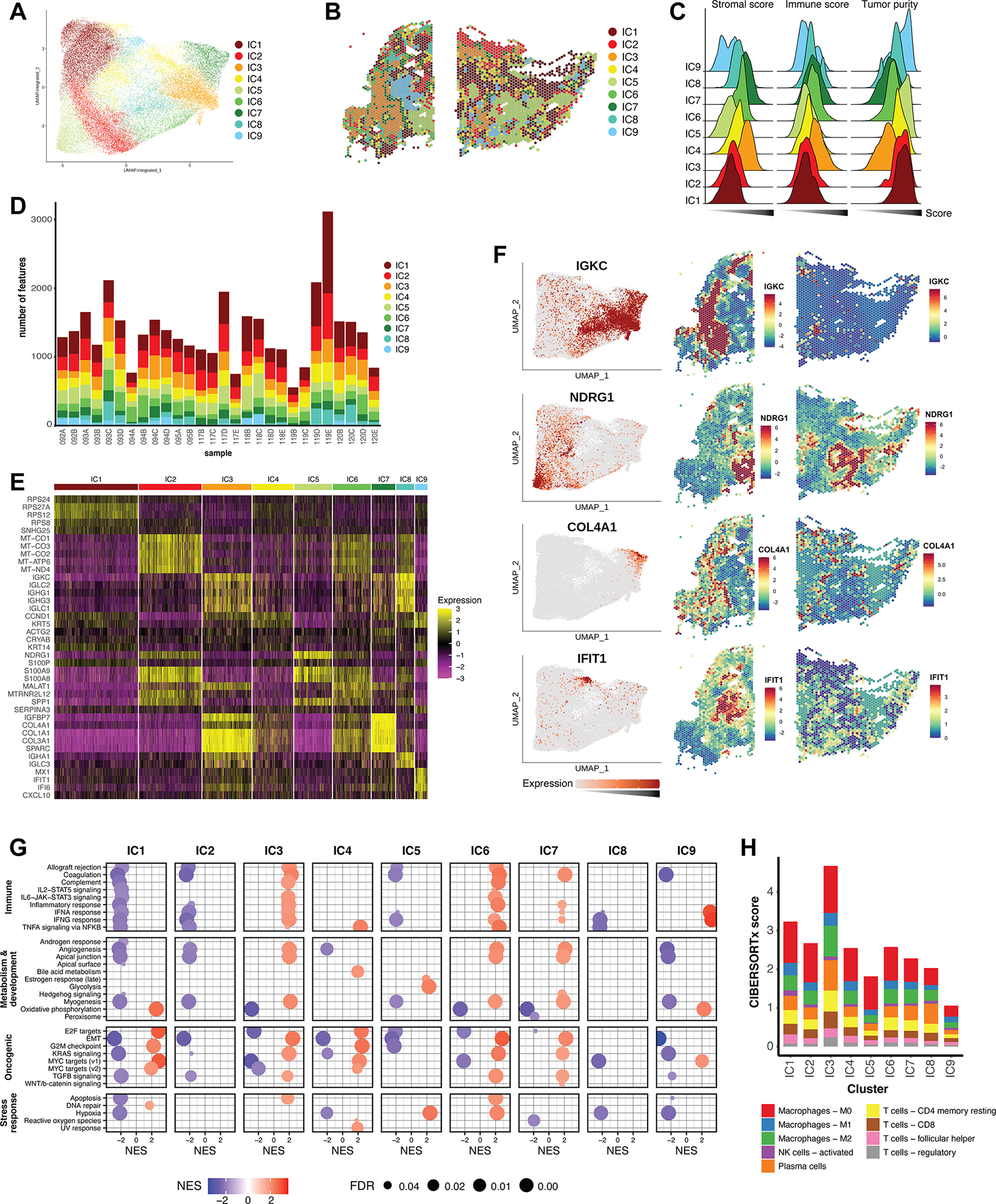
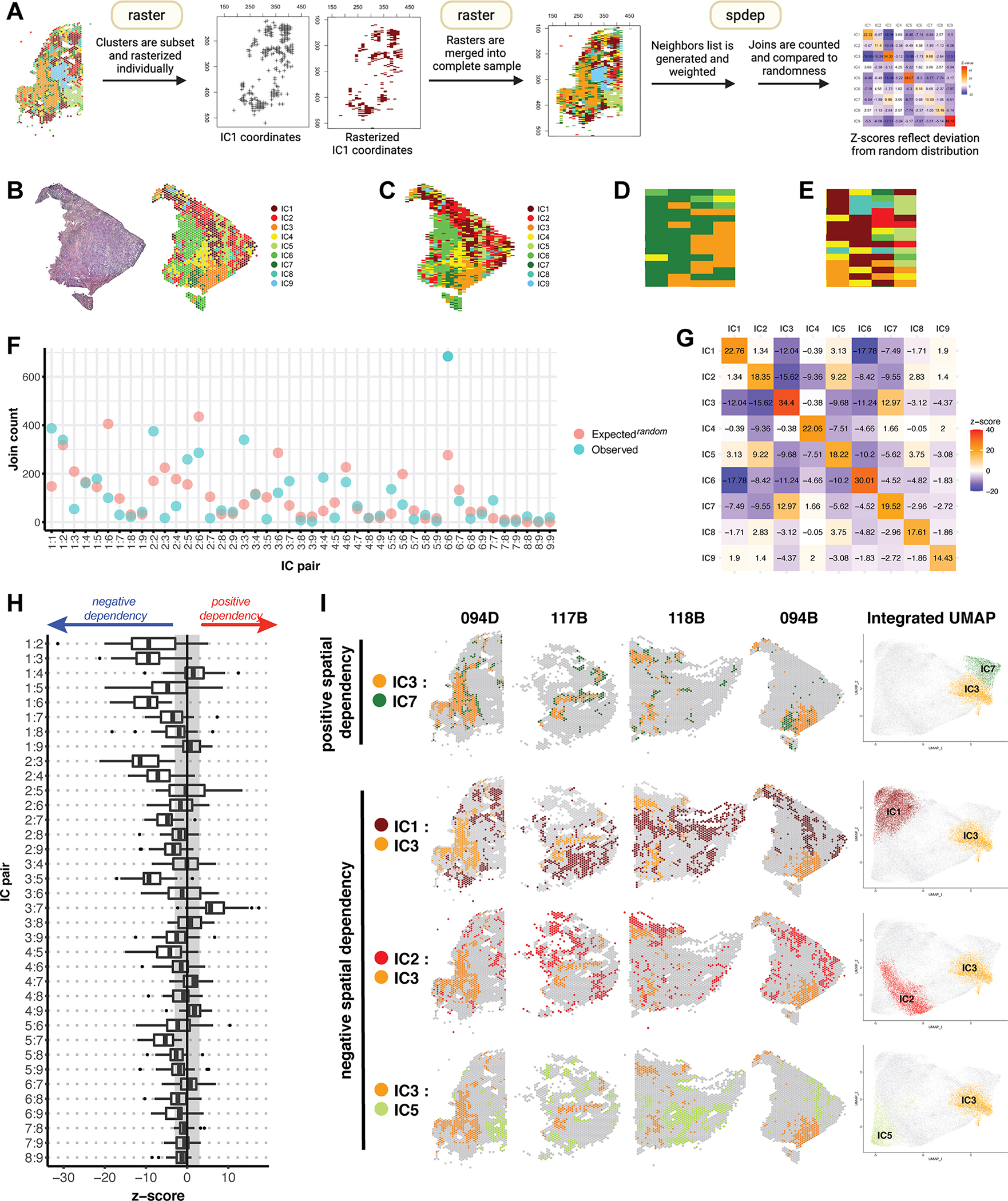
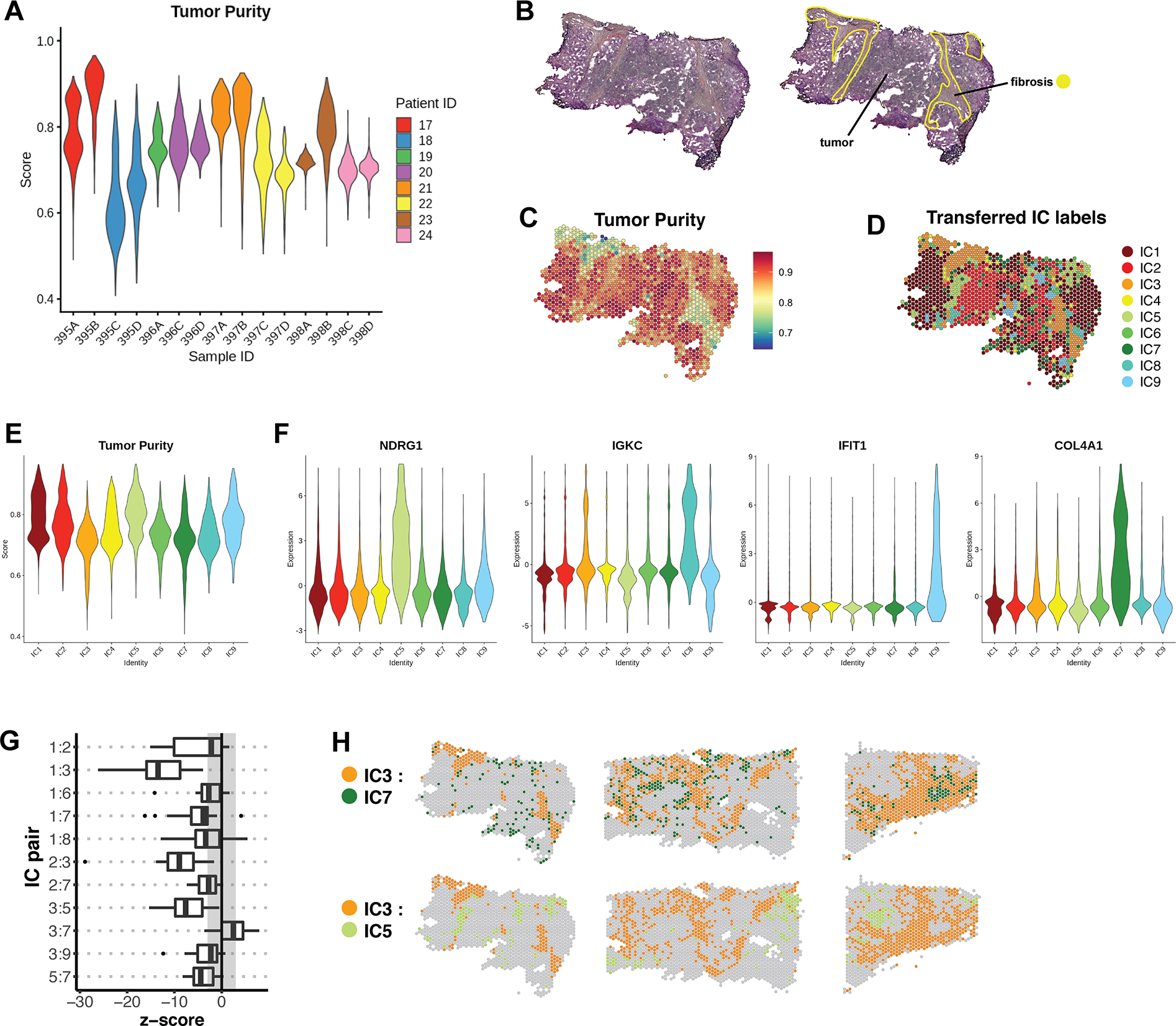
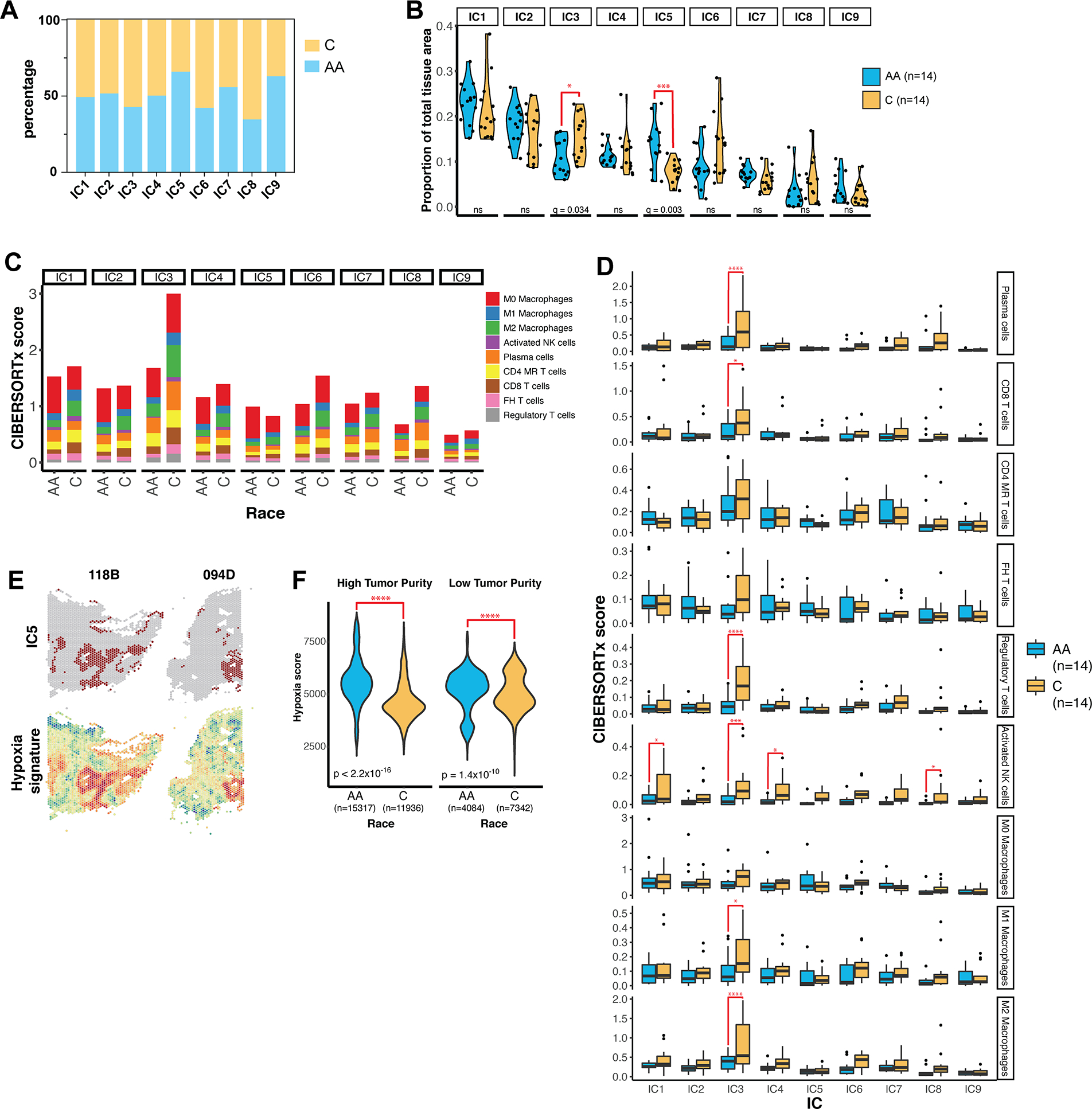
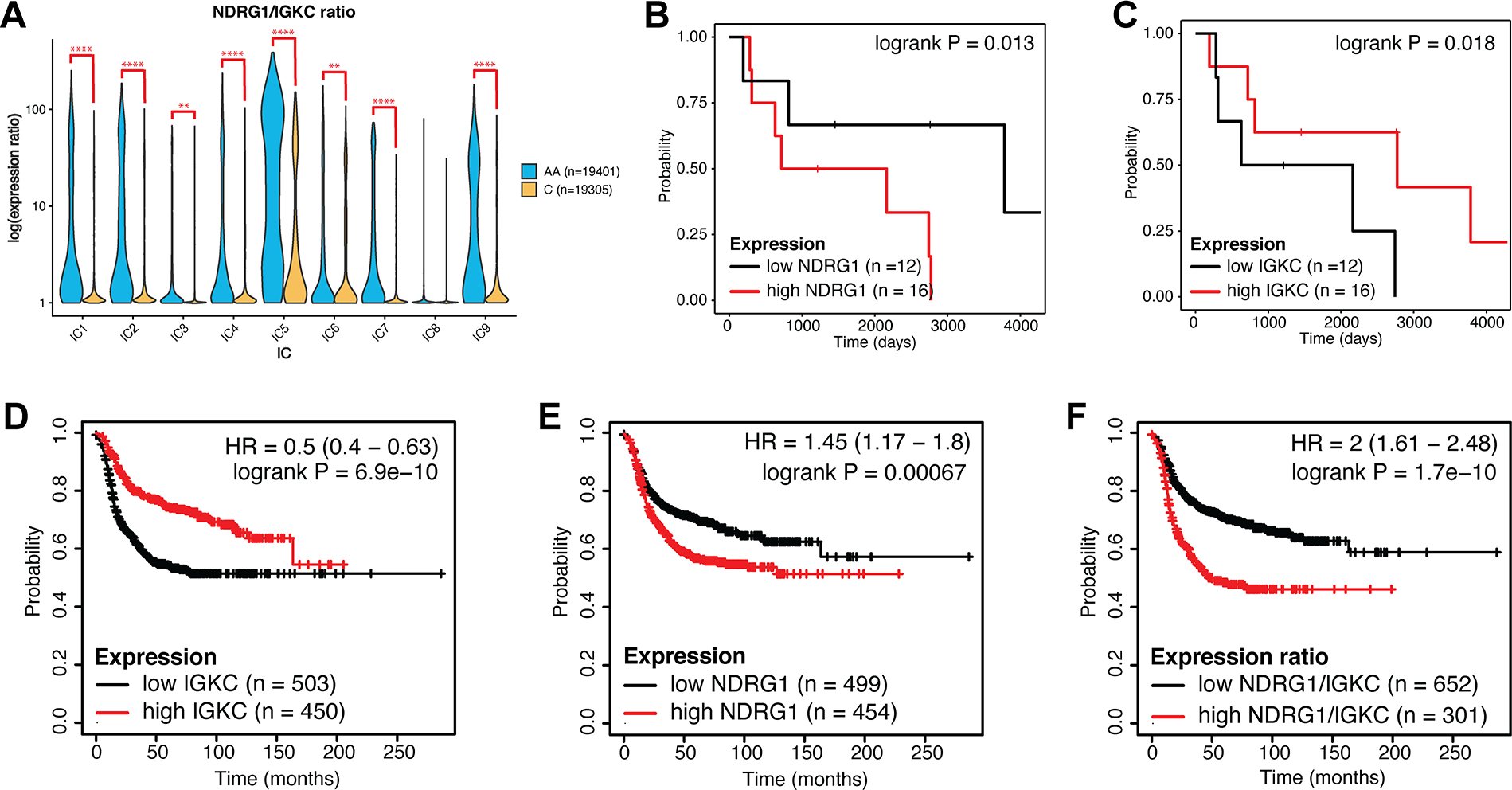
Triple-negative breast cancer (TNBC) is an aggressive disease that disproportionately affects African American (AA) women. Limited targeted therapeutic options exist for patients with TNBC. Here, we employ spatial transcriptomics to interrogate tissue from a racially diverse TNBC cohort to comprehensively annotate the transcriptional states of spatially resolved cellular populations. A total of 38,706 spatial features from a cohort of 28 sections from 14 patients were analyzed. Intratumoral analysis of spatial features from individual sections revealed heterogeneous transcriptional substructures. However, integrated analysis of all samples resulted in nine transcriptionally distinct clusters that mapped across all individual sections. Furthermore, novel use of join count analysis demonstrated nonrandom directional spatial dependencies of the transcriptionally defined shared clusters, supporting a conserved spatio-transcriptional architecture in TNBC. These findings were substantiated in an independent validation cohort comprising 17,861 spatial features representing 15 samples from 8 patients. Stratification of samples by race revealed race-associated differences in hypoxic tumor content and regions of immune-rich infiltrate. Overall, this study combined spatial and functional molecular analyses to define the tumor architecture of TNBC, with potential implications in understanding TNBC disparities.
Significance: Spatial transcriptomics profiling of a diverse cohort of triple-negative breast cancers and innovative informatics approaches reveal a conserved cellular architecture across cancers and identify proportional differences in tumor cell composition by race.
©2022 American Association for Cancer Research.
Conflict of interest statement
Figures
References
-
- Foulkes WD, Smith IE, Reis JS. Triple-Negative Breast Cancer. New Engl J Med. 2010;363(20):1938–48. - PubMed
-
- Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13(15 Pt 1):4429–34. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
