

Bi-allelic CAMSAP1 variants cause a clinically recognizable neuronal migration disorder
- PMID: 36283405
- PMCID: PMC9674946
- DOI: 10.1016/j.ajhg.2022.09.012
Bi-allelic CAMSAP1 variants cause a clinically recognizable neuronal migration disorder
Abstract
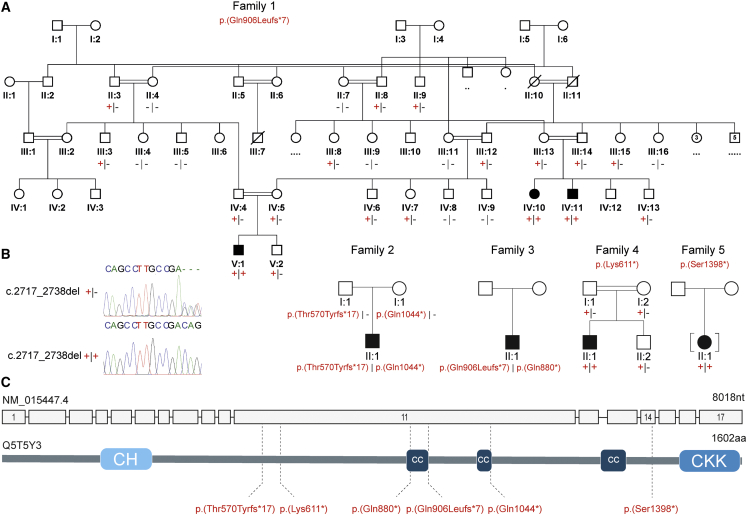
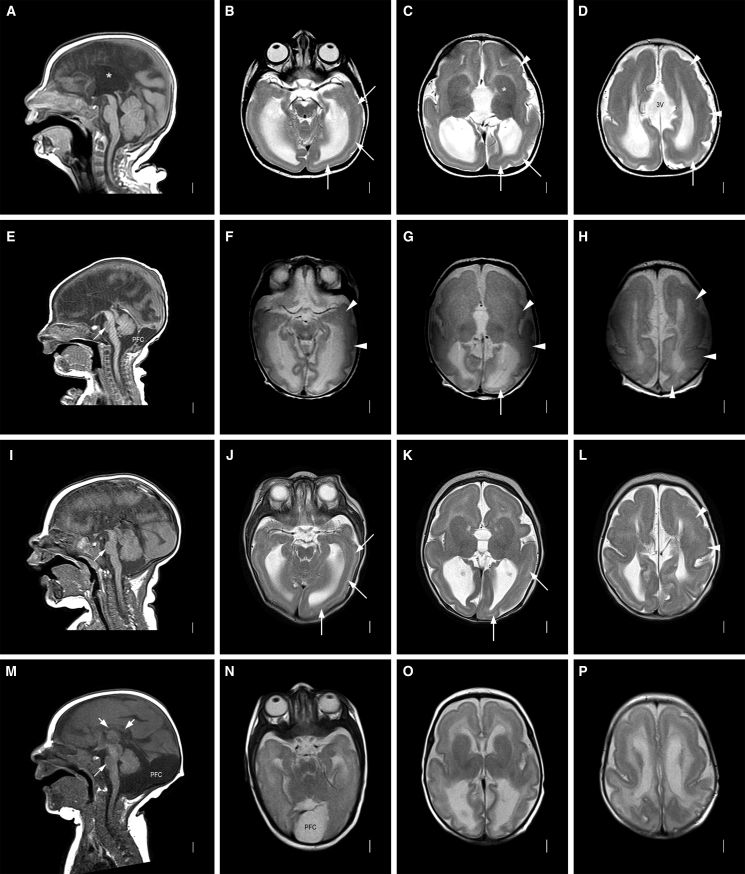
Non-centrosomal microtubules are essential cytoskeletal filaments that are important for neurite formation, axonal transport, and neuronal migration. They require stabilization by microtubule minus-end-targeting proteins including the CAMSAP family of molecules. Using exome sequencing on samples from five unrelated families, we show that bi-allelic CAMSAP1 loss-of-function variants cause a clinically recognizable, syndromic neuronal migration disorder. The cardinal clinical features of the syndrome include a characteristic craniofacial appearance, primary microcephaly, severe neurodevelopmental delay, cortical visual impairment, and seizures. The neuroradiological phenotype comprises a highly recognizable combination of classic lissencephaly with a posterior more severe than anterior gradient similar to PAFAH1B1(LIS1)-related lissencephaly and severe hypoplasia or absence of the corpus callosum; dysplasia of the basal ganglia, hippocampus, and midbrain; and cerebellar hypodysplasia, similar to the tubulinopathies, a group of monogenic tubulin-associated disorders of cortical dysgenesis. Neural cell rosette lineages derived from affected individuals displayed findings consistent with these phenotypes, including abnormal morphology, decreased cell proliferation, and neuronal differentiation. Camsap1-null mice displayed increased perinatal mortality, and RNAScope studies identified high expression levels in the brain throughout neurogenesis and in facial structures, consistent with the mouse and human neurodevelopmental and craniofacial phenotypes. Together our findings confirm a fundamental role of CAMSAP1 in neuronal migration and brain development and define bi-allelic variants as a cause of a clinically distinct neurodevelopmental disorder in humans and mice.
Keywords: MARK2; agyria; autosomal recessive; lissencephaly; neurodevelopmental disorder; pachygyria; patronin; tubulinopathy.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures


References
-
- Fry A.E., Cushion T.D., Pilz D.T. The genetics of lissencephaly. Am. J. Med. Genet. C Semin. Med. Genet. 2014;166C:198–210. - PubMed
-
- Smith D.S., Niethammer M., Ayala R., Zhou Y., Gambello M.J., Wynshaw-Boris A., Tsai L.H. Regulation of cytoplasmic dynein behaviour and microtubule organization by mammalian Lis1. Nat. Cell Biol. 2000;2:767–775. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
